Kamagra repose sur le sildénafil comme principe actif, avec un mode d’action identique à celui du Viagra. La forme galénique en gel oral permet une absorption plus rapide et une concentration plasmatique maximale plus précoce que les comprimés. Le mécanisme implique l’inhibition compétitive de la PDE5, entraînant une relaxation musculaire lisse locale et une vasodilatation ciblée. La demi-vie courte, environ 4 heures, limite la durée d’action. L’élimination se fait après métabolisme hépatique, impliquant majoritairement le CYP3A4. L’incidence d’effets indésirables comprend céphalées, rougeurs et congestion nasale, de façon transitoire. Dans les comparatifs pharmacologiques, acheter kamagra sans ordonnance est associé aux présentations galéniques alternatives disponibles.
Port.duke-nus.edu.sg
Basic Science Reports Non-Equilibrium Gating in Cardiac Na؉ Channels An Original Mechanism of Arrhythmia
Colleen E. Clancy, PhD; Michihiro Tateyama, PhD; Huajun Liu, MD;
Xander H.T. Wehrens, MD, PhD; Robert S. Kass, PhD
Background—Many long-QT syndrome (LQTS) mutations in the cardiac Naϩ channel result in a gain of function due to
a fraction of channels that fail to inactivate (burst), leading to sustained current (Isus) during depolarization. However,some Naϩ channel mutations that are causally linked to cardiac arrhythmia do not result in an obvious gain of functionas measured using standard patch-clamp techniques. An example presented here, the SCN5A LQTS mutant I1768V,does not act to increase Isus (Ͻ0.1% of peak) compared with wild-type (WT) channels. In fact, it is difficult to reconcilethe seemingly innocuous kinetic alterations in I1768V as measured during standard protocols under steady-stateconditions with the disease phenotype. Methods and Results—We developed new experimental approaches based on theoretical analyses to investigate Naϩ
channel gating under non-equilibrium conditions, which more closely approximate physiological changes in membranepotential that occur during the course of a cardiac action potential. We used this new approach to investigatechannel-gating transitions that occur subsequent to channel activation. Conclusions—Our data suggest an original mechanism for development of LQT-3 arrhythmias. This work demonstrates
that a combination of computational and experimental analysis of mutations provides a framework to understand complex mechanisms underlying a range of disorders, from molecular defect to cellular and systems function. (Circulation. 2003;107:2233-2237.) Key Words: arrhythmia Ⅲ remodeling Ⅲ sodium Ⅲ long-QT syndrome
Ionchannelsareadiversegroupofpore-formingtransmem- which ventricular repolarization is prolonged.8 Investigation
brane proteins that selectively conduct ions and play
of the disease-associated mutant channels revealed defects in
physiological roles in most cell types, including neurons,
channel inactivation such that during the prolonged plateau
skeletal muscle, smooth muscle, and cardiac muscle. Inher-
phase of the cardiac ventricular action potential, a small
ited mutations in genes encoding ion channels have been
number of channels reopen and conduct Naϩ ions instead of
associated with such a large number of human diseases,
entering an absorbing non-conducting inactivated state, cre-
including epilepsy, febrile seizures, Dent’s disease, and
ating sustained Naϩ current (Isus).9 This mutation-altered
cardiac arrhythmias, that the disorders are called “chan-
channel function was demonstrated in computational models
nelopathies.”1–7 Expression of ion channels in heterologous
and in genetically altered mice to account for the disease
systems allows for investigation of inherited ion channel
phenotype.10,11 Recently, mutations in SCN1A, the gene
defects at the single protein and cellular level to directly
coding for the human neuronal Naϩ channel ␣-subunit, that
identify the disease-associated alterations in ion channel
are associated with epilepsy have been reported to cause
function. Disease-linked mutations provide an opportunity to
similar defects in channel inactivation gating and promotion
understand the mechanistic basis of human disease, from
of Isus.5 As of yet, the cellular consequences of such epilepsy
altered molecular function to the clinical syndrome. This
mutations remain elusive. Thus, mechanistic insights gained
approach has led to novel insight into roles of key ion
from investigation of cardiac defects are likely to have
channels in human physiology and pathophysiology.
Perhaps one of the most unexpected and interesting reve-
However, not all LQT-3 mutations in SCN5A cause this
lations is the link between mutations in SCN5A, the gene
type of altered channel behavior, and understanding how
coding for the ␣-subunit of the cardiac Naϩ channel, and
these other mutations cause the disease phenotype, mani-
variant 3 of the long-QT syndrome (LQT-3), a disease in
fested as prolongation of the ECG QT interval, has not been
Received February 24, 2003; revision received March 13, 2003; accepted March 13, 2003. From the Department of Pharmacology, Columbia University College of Physicians and Surgeons, New York, NY. The online-only Data Supplement is available at http://www.circulationaha.org. This article originaly appeared Online on April 14, 2003 (Circulation. 2003;107:r70 –r74). Correspondence to Robert S. Kass, PhD, Department of Pharmacology, Columbia University College of Physicians and Surgeons, 630 W 168th St,
New York, NY 10032. E-mail [email protected]
2003 American Heart Association, Inc. Circulation is available at http://www.circulationaha.org DOI: 10.1161/01.CIR.0000069273.51375.BD 2233 2234 Circulation May 6, 2003
obvious based on the analysis of mutant channel biophysicalproperties. Examples of mutations that do not result in gain offunction include D1790G, E1295K, and I1768V.12–14 Thesedefect types led us to ask, how might mutations that havesubtle effects on channel kinetics underlie severe patientphenotypes? Can we investigate Naϩ channel gating differ-ently to reveal arrhythmia cellular mechanisms and alteredchannel function that might be relevant to other diseases?
The approach we have taken was to use computer model-
ing to first analyze theoretical and subtle changes in channel
Diagram. A schematic of the voltage protocol. Persistent late
gating that might underlie the disease phenotype, but which
current Isus was measured after 100 ms depolarization to ϩ20mV, indicated by ➀ in the diagram. Ramp currents (Iramp) were
may have been overlooked in previous experimental investi-
measured as the peak inward current during the negative ramp
gations. We utilized a theoretical cardiac Naϩ channel mod-
el10,15 to guide our experimental approach to investigategenetic defects.16–18 The model suggested that mutation-
Electrophysiology
altered gating transitions subsequent to channel activation,
Membrane currents were measured using whole cell patch-clamp
driven by changes in membrane potential during repolariza-
procedures, with Axopatch 200B amplifiers (Axon Instruments). Capacity current and series resistance compensation were carried out
tion, might determine action potential duration. Because
using analog techniques according to the amplifier manufacture
recovery from open-state inactivation is time- and voltage-
(Axon Instruments). All measurements were obtained at room
dependent, standard voltage clamp protocols may fail to
temperature (22°C). Macroscopic whole cell Naϩ current was rec-
reveal mutation-induced changes in kinetics that exist under
orded using the following solutions (mmol/L). The internal solution
conditions in which voltage changes.
contained aspartic acid 50, CsCl 60, Na -ATP 5, EGTA 11, HEPES
10, CaCl 1, and MgCl 1, with pH 7.4 adjusted with CsOH. The
We chose to focus on the I1768V LQT3 mutation in the
external solution contained NaCl 130, CaCl 2, CsCl 5, MgCl 1.2,
cardiac Naϩ channel because a previous study that investi-
HEPES 10, and glucose 5, with pH 7.4 adjusted with CsOH. Using
gated alterations in steady state channel gating revealed that
WEBMAXCLITE v1.15,22 at 22°C at pH 7.4 and ionic strength ϭ
the mutation sped recovery from inactivation.13 In that study,
0.16N, we computed the free Ca2ϩ and Mgϩ as 6.888e-9 mol/L and
a long slow (steady-state) positive ramp protocol also re-
0.0000245 mol/L, respectively. The voltage protocols are describedin the text. Negative ramp currents were measured as tetrodotoxin
vealed subtle changes in window current, which were sug-
(TTX) -sensitive current by applying TTX at high concentrations (30
gested as a potential arrhythmia mechanism. In the present
mol/L) to block expressed Naϩ channel currents and reveal
study, our computational analysis led us to believe that faster
background currents, which were then subtracted digitally. A sche-
recovery from open state inactivation in I1768V may be the
matic of the voltage protocol is shown in the Diagram. Persistent latecurrent I
was measured after 100 ms depolarization to ϩ20 mV,
major factor in determining disease phenotype.
indicated by ① in the diagram. Ramp current (I
We provide experimental evidence in support of this
the peak inward current during the negative ramp (indicated as ② in
hypothesis and propose that mutation-induced gain of func-
the Diagram). Holding potentials were Ϫ100 mV. Analysis was
performed in Excel (Microsoft) and Origin 6.1 (Microcal Software).
distinct forms. The most common is due to transient inacti-
Data are represented as meanϮSEM. Statistical significance wasdetermined using unpaired Student’s t test; PϽ0.05 was considered
vation failure, termed bursting, which underlies sustained
Naϩ channel activity over the plateau voltage range.9,19 Asecond is due to steady-state channel reopening called win-
Computational Methods
dow current,20 because reopening occurs over voltage ranges
All computational methods have been described in previous publi-
for which steady-state inactivation and activation overlap.
cations.10,15 Action potentials were computed by incorporating this
Here we demonstrate a third original mechanism that occurs
into a previously described cellular model.15
under non-equilibrium conditions whereby channel reopening
Model Framework
results from faster recovery from inactivation at membrane
Mutant channels differ from WT channels in one distinct way, as
potentials that facilitate the activation transition. We find that
evidenced by experimental recordings. Mutant channels have altered
mutation induced faster recovery from inactivation results in
rates of recovery from channel inactivation due to faster rates ofrecovery from channel inactivation (Data Supplement). The faster
channels that reopen during repolarization and that the
recovery from inactivation is simulated in mutant I1768V channels
resulting current amplitude rivals that of bursting channels.
by doubling the rates of recovery from inactivation (UIM2 3 UIM1,
Using the Luo-Rudy virtual transgenic cell,21 we demonstrate
UIM1 3 UIF, UIC3 3 UC3, UIC2 3 UC2, UIF 3 UC1)
that late current due to channels reopening causes severe
prolongation of the AP plateau and arrhythmic triggers.
model contains 2 possible modes of gating, a “background mode”and a “burst mode.” The background mode includes the upper 9
states, which consist of 3 closed states (UC3, UC2, UC1), aconducting open state (UO), a fast inactivation state (UIF), and 2
Expression of Recombinant Na؉ Channels
intermediate inactivation states (UIM1 and UIM2) that are required
Naϩ channels were expressed in human embryonic kidney 293 cells
to reproduce the complex fast and slow recovery features of
at 22°C as described previously.14 CD8-positive cells identified
inactivation. Channels enter the IM2 state via slow transitions.
using Dynabeads (Dynal, M-450) were patch clamped 48 hours after
Channel closed-state inactivation is achieved via the inclusion of 2
closed inactivation states (UIC2 and UIC3). The lower four states
Clancy et al Non-Equilibrium Gating in Cardiac Na؉ Channels 2235 Figure 1. The I1768V mutation (right) has little effect on whole cell currents compared with wild-type (left) in experiments (top, line indicates zero current level) and simulations (bottom). Figure 2. A, Non-equilibrium gating. Simulated (left) and experi- mental (right) macroscopic currents during the negative ramp
(prefixed with L, denoting “lower”) correspond to a burst mode of
protocol (see text). B, Summary of Isus (plotted as % peak cur-
gating that corresponds to channels that lack inactivation. This
rent, measured at time indicated by arrows in A) after 100 ms
population is unchanged by the I1769V mutation and is negligible
depolarization to ϩ20 mV (Isus, reflects bursting channels) and
but included for accuracy in both WT and I1768V mutant channels
peak current during the ramp repolarization (plotted as % peak
(Ͻ0.07% of peak current after 100 ms depolarization to ϩ20 mV).
current) (Iramp, reflects channel reopenings). There is no signifi-
Transition rates between upper and lower states represent a proba-
cant difference between Isus WT and Isus I1768V. For Iramp, there
bility of transition between the 2 modes of gating. Microscopic
is a significant difference in maximum ramp current between WT
reversibility was ensured by fixing the products of the forward and
and I1768V channels. Number of experiments: nϭ6, WT; nϭ10,IV. * Pϭ0.02.
reverse transition rates in closed loops of the model.
All the simulations were encoded in C/Cϩϩ. Simulations were
implemented (double precision) on an Apple Macintosh 500 mHz G4
between Isus in WT (Ϫ0.39A/F, 0.07%) or I1768V
Powerbook (Motorola) running OS X. A time step of 0.005 ms was
(Ϫ0.40A/F, 0.07%) simulated cells (Figure 2A) was noted.
used.23 Computer code used for computations in this paper is
The arrows in Figure 2A indicate the end of the 100 ms
available on request by e-mailing [email protected].
depolarization, when Isus was measured. Summarized data are
shown as percentages of peak current in Figure 2B, left.
The effects of the I1768V mutation on steady-state currents
Consistent with the computations, we found that I1768V
were previously reproduced by incorporating a 2-fold in-
mutants expressed in human embryonic kidney 293 cells
crease in the rate of recovery from channel inactivation in a
exhibit larger transient inward current (Ϫ0.33A/F, 0.18% of
computer model of cardiac Naϩ channel current (INa) (Data
peak current [Ϫ202.1A/F]) during repolarization than WT
Supplement).15 Faster recovery from inactivation had no
channels (Ϫ0.31A/F, 0.12% of peak current [Ϫ259.1A/F)
effect on current density, sustained current (channel burst-
(Figure 2A) with no change in bursting, because late current
ing), activation (not shown), or the voltage-dependence and
measured after 100 ms depolarization to Ϫ20 mV (arrow in
time course of currents activated during depolarization (Fig-
Figure 2A) is identical in WT (Ϫ0.14A/F, 0.05%) and
ure 1, lower), consistent with experimental data (Figure 1,
I1768V (Ϫ0.13A/F, 0.06%) channels. Again, the arrows
indicate the end of the 100 ms depolarization when Isus was
We next investigated non-equilibrium gating of WT and
measured. Importantly, the larger current is not window
I1768V channels using a theoretical and then an experimental
current, as the voltage of peak Iramp occurs outside the overlap
approach. We computed currents (Iramp) using a negative ramp
of activation and inactivation (WT peak Iramp occurs at Ϫ24.17
protocol. In the computation, cells first were depolarized
(ϩ20 mV for 100 ms from holding potential [V
Summary data for experimentally determined I
mV) to promote open state inactivation. As noted previously,
presented in Figure 2B, right panel. The experimental result is
at this voltage, after opening, channels enter an absorbing
nearly identical to the theoretical simulation; I
inactivated state and there is very little sustained current
for WT and IV channels (0.05Ϯ0.01%, nϭ6; and
(measured after 100 ms at ϩ20 mV, indicated by arrows in
0.07Ϯ0.07%, nϭ6, not significant, respectively), whereas
Figure 2A). Gradual repolarization was then applied over 100ms until V ϭϪ
Iramp is significantly increased (0.12Ϯ0.006%, nϭ6; and
100 mV, allowing for recovery from inacti-
vation. We first used the model to investigate the conse-
0.17Ϯ0.02%, nϭ10, Pϭ0.02, respectively).
quences of an increased rate of recovery from inactivation on
It should be noted that the larger Iramp observed in I1768V
cells is not attributable to an increase in driving force because
the small fraction of current that remains at the end of the 100
increase in the channel recovery rate results in nearly a
ms depolarization (Isus) is present in both the WT- and mutant
channel-containing cells. It is also not due to longer openings
ramp for the I1768V mutation (Ϫ1.23A/F, 0.2% of peak
in mutant channels, as the time course of the macroscopic
current [Ϫ581.6A/F]) compared with WT (Ϫ0.738A/F, 0.1%
current decay was not affected by the mutation, and previous
of peak current [Ϫ580.0A/F]) (Figure 2A), but no difference
measurements of single channels reveal identical gating.13
2236 Circulation May 6, 2003
phenotype. By using a computational analysis of ion channelactivity, we developed targeted experiments to dissect subtlechanges in channel gating that, within the framework of thecomputational model, were capable of causing the diseasephenotype. A great advantage of analyzing the relationshipbetween inherited defects in cardiac ion channels and theclinical disorders they cause is the fact that the electricalcharacteristics of the disease phenotype can be measureddirectly (via the ECG) and compared with the expectedchanges in cellular function caused by the experimentally
Figure 3. The I1768V mutation disrupts cellular repolarization in
determined alteration in channel function. Another advan-
a rate-dependent manner. Faster recovery from inactivation
tage, which we demonstrate in the current study study, is the
resulting from the I1768V mutation results in channel reopenings
utility of computational models of both cellular and ion
during repolarization of the AP (19th and 20th paced APs),which prolongs the APD at a pacing rate of 1200 ms (A) and
channel electrophysiology that have been developed for
leads to arrhythmogenic early afterdepolarizations as the rate is
In the present study, we demonstrate a third and novel
mechanism by which mutations in the cardiac Naϩ channel
Is this single change in kinetics sufficient to disrupt cellular
may lead to a gain of channel function that leads to Naϩ
repolarization? We tested I1768V mutant channels in the
current during the action potential plateau. The most common
Luo-Rudy model of the cardiac action potential.21 Effects of
gain of function defect is due to transient failure of channel
the I1768V (red line) mutation on cellular repolarization
inactivation, a mode of gating termed bursting, which under-
compared with WT (black line) at 3 pacing rates are shown in
lies sustained Naϩ channel activity over the plateau voltage
Figure 3 (A: 1200 ms, B: 1500 ms, and C: 2000 ms). The AP
range.9,19 A second mechanism results from steady-state
simulations reveal that the I1768V mutation disrupts cellular
channel reopening, called window current,20 because reopen-
repolarization in a rate-dependent manner as described pre-
ing occurs over voltage ranges for which steady-state inacti-
viously in the clinical phenotype.13 As the pacing rate is
vation and activation overlap. Here, we demonstrate a third
progressively slowed (Figure 3B and 3C), the I1768V muta-
original mechanism that occurs under non-equilibrium con-
tion results in formation of arrhythmogenic early afterdepo-
ditions whereby channel reopening results from faster recov-
ery from inactivation at membrane potentials that facilitate
The mechanism of I1768V disruption of cellular repolar-
the activation transition. This third type of gain of function
ization is shown in Figure 4. The 19th and 20th WT (left) and
can be distinguished from window current by considering the
I1768V (right) APs after pacing at CLϭ2000 ms are shown
voltage (Ϫ20 mV) at which the reopening occurs, which is
with corresponding INa at high gain. Clearly, the I1768V
outside of the region of overlap of the activation and
mutation results in a much larger inward current (arrows)
inactivation curves. It should be noted that the population of
compared with WT, because of faster recovery of Naϩ
channels that recover at plateau membrane potentials repre-
channels from inactivation and subsequent channel reopen-
sents a tiny fraction of the channel population that recover
ing. The reopenings result in a relatively large INa during the
more rapidly because of the channel mutation. Although
normally delicately controlled AP plateau. Indeed, the current
recovery at plateau potentials is generally unfavorable, the
amplitude is at least as large as that observed for the ⌬KPQ
mutation increases the propensity of channels to reopen under
mutation, known to result in severe patient phenotypes.24
non-equilibrium conditions, ie, during changing voltage, that
Moreover, the repolarization rate during the AP seems to
are not obvious during steady-state voltage protocols. We
exacerbate the channel reopenings, thereby resulting in stable
find that mutation induced faster recovery from inactivation
results in channels that reopen during repolarization and thatthe resulting current amplitude rivals that of bursting chan-
Discussion
nels. Using a virtual transgenic cell,21 we demonstrate that
Here we have used a novel approach to elucidate the link
late current due to channels reopening causes severe prolon-
between an inherited ion channel mutation and its disease
gation of the AP plateau and arrhythmic triggers.
It is notable that the time-course of late INa is different
during the action potential (Figure 4) compared with thatobserved during the negative ramp (Figure 2A). There areseveral things that make the current morphology different. First, the negative ramp protocol begins with a 100 msdepolarization to 20 mV, which was chosen deliberately toobserve channel transitions that occur subsequent to channelopen-state inactivation during the plateau phase of the actionpotential. The long depolarization to force channel inactiva-
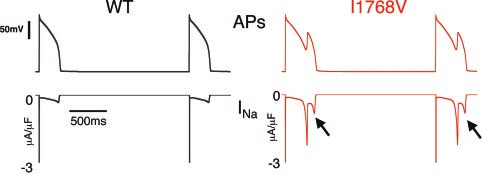
Figure 4. Mechanism of abnormal repolarization. The 19th and
tion results in a pseudo steady-state open channel inactiva-
20th paced (rateϭ2000 ms) WT (left) and I1768V (right) action
tion, which allows for the unencumbered study of channel
potentials are shown with corresponding INa at high gain shownbeneath. Clancy et al Non-Equilibrium Gating in Cardiac Na؉ Channels 2237
In the case of the action potential, the membrane potential
4. Schwake M, Friedrich T, Jentsch TJ. An internalization signal in ClC-5,
is constantly changing, thereby making it difficult to deter-
an endosomal Cl-channel mutated in Dent’s disease. J Biol Chem. 2001;276:12049 –12054.
mine exactly which transitions are occurring at any given
5. Lossin C, Wang DW, Rhodes TH, et al. Molecular basis of an inherited
time. Indeed, it appears that the rate of change of the
epilepsy. Neuron. 2002;34:877– 884.
membrane potential during the action potential promotes
6. Jurkat-Rott K, Lehmann-Horn F. Human muscle voltage-gated ion
channel reopening even more than that observed during the
channels and hereditary disease. Curr Opin Pharmacol. 2001;1:280 –287.
7. Kullmann DM. The neuronal channelopathies. Brain. 2002;125:
negative ramp. This occurs because the action potential
upstroke is followed by immediate repolarization. Hence,
8. Wang Q, Shen J, Splawski I, et al. SCN5A mutations associated with an
fewer channels enter intermediate inactivation states, increas-
inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805– 811.
ing the likelihood of reopening. In Figure 4, the I1768V Naϩ
9. Bennett PB, Yazawa K, Makita N, et al. Molecular mechanism for an
inherited cardiac arrhythmia. Nature (Lond). 1995;376:683– 685.
current exhibits 2 late Naϩ peaks during AP repolarization.
10. Clancy CE, Rudy Y. Linking a genetic defect to its cellular phenotype in
The first peak (2.2A/F) occurs at Ϫ21 mV, and the second
a cardiac arrhythmia. Nature (Lond). 1999;400:566 –569.
(0.8A/F) occurs at Ϫ35 mV. The occurrence of the second
11. Nuyens D, Stengl M, Dugarmaa S, et al. Abrupt rate accelerations or
premature beats cause life-threatening arrhythmias in mice with
Na peak is due to a different mechanism than the first.
long-QT3 syndrome. Nat Med. 2001;7:1021–1027.
The first peak is due to mutation-induced faster channel
12. An RH, Wang XL, Kerem B, et al. Novel LQT-3 mutation affects Naϩ
recovery from inactivation and then re-inactivation as the
channel activity through interactions between alpha- and beta1-subunits.
secondary depolarization, carried by Ca2ϩ, results in more
positive membrane potentials that favor inactivation. The
13. Rivolta I, Clancy CE, Tateyama M, et al. A novel SCN5A mutation
associated with long QT-3: altered inactivation kinetics and channel
second late INa peak is much smaller and occurs because of
dysfunction. Physiol Genomics. 2002;10:191–197.
reactivated channels that are again recovering from inactiva-
14. Abriel H, Cabo C, Wehrens XH, et al. Novel arrhythmogenic mechanism
tion. In this case, the repolarizing membrane is less favorable
revealed by a long-QT syndrome mutation in the cardiac Na(ϩ) channel.
to inactivation and channels reopen before deactivating in
Circ Res. 2001;88:740 –745.
15. Clancy CE, Rudy Y. Na(ϩ) channel mutation that causes both Brugada
and long-QT syndrome phenotypes. a simulation study of mechanism.
Here we show that this computational approach allowed us
Circulation. 2002;105:1208 –1213.
to determine subtle changes in channel gating that previously
16. Noble D. Modeling the heart: from genes to cells to the whole organ.
had not been investigated within the context of alteration in
Science. 2002;295:1678 –1682.
17. Noble D. The rise of computational biology. Nat Rev Mol Cell Biol.
cellular repolarization. It must be noted, however, that mod-
eling of complex biological processes is not without limita-
18. Noble D. Unraveling the genetics and mechanisms of cardiac arrhythmia.
tions. By definition, a model is a simplification of the actual
Proc Natl Acad Sci U S A. 2002;99:5755–5756.
biological process that allows for insight and understanding
19. Chandra R, Starmer CF, Grant AO. Multiple effects of KPQ deletion
mutation on gating of human cardiac Naϩ channels expressed in mam-
but may result in the exclusion of details necessary for
malian cells. Am J Physiol. 1998;274:H1643–H1654.
absolute understanding of biological complexity and mecha-
20. Wang DW, Yazawa K, George ALJ, et al. Characterization of human
nism. Nonetheless, the combination of theoretical prediction
cardiac Naϩ channel mutations in the congenital long QT syndrome.
and experimental verification has led to the identification of
Proc Natl Acad Sci U S A. 1996;93:13200 –13205.
21. Luo CH, Rudy Y. A dynamic model of the cardiac ventricular action
a novel mechanism through which altered Naϩ channel
potential: I: simulations of ionic currents and concentration changes. Circ
activity can account for prolonged QT intervals in mutation
carriers. Although the waveform of the cardiac action poten-
22. Patton C. WEBMAXCLITE v1.15. Available at: http://www.
tial and the electrical properties that define its plateau phase
stanford.edu/~cpatton/webmaxclite115.htm. Accessed March 27, 2003.
23. Cardiac Bioelectricity Research and Training Center. Research. Available
are unique, this integrative approach is applicable to under-
at: http://www.cwru.edu/med/CBRTC/Research.html. Accessed March
standing the molecular basis of other congenital diseases,
such as myotonia and epilepsy, in which subtle changes in
24. Schwartz PJ, Priori SG, Spazzolini C, et al. Genotype-phenotype corre-
Naϩ channel gating may increase the contribution of channel
lation in the long-QT syndrome: gene-specific triggers for life-threateningarrhythmias. Circulation. 2001;103:89 –95.
reopenings to myotonic discharge (myotonias)25–28 or bursts
25. Wang DW, VanDeCarr D, Ruben PC, et al. Functional consequences of
of neural activity (epilepsy and seizure disorders).5,29
a domain 1/S6 segment sodium channel mutation associated with painfulcongenital myotonia. FEBS Lett. 1999;448:231–234. Acknowledgments
26. Jurkat-Rott K, Mitrovic N, Hang C, et al. Voltage-sensor sodium channel
mutations cause hypokalemic periodic paralysis type 2 by enhanced
This work was supported by grants 1R01-HL 56810-5 and 1P01-HL
inactivation and reduced current. Proc Natl Acad Sci U S A. 2000;97:
67849-02 from the National Institutes of Health, Heart, Lung and
27. Desaphy JF, De Luca A, Tortorella P, et al. Gating of myotonic Na
channel mutants defines the response to mexiletine and a potent
References
derivative. Neurology. 2001;57:1849 –1857.
1. Marban E. Cardiac channelopathies. Nature. 2002;415:213–218.
28. Green DS, George AL Jr, Cannon SC. Human sodium channel gating
2. Jentsch TJ. Neuronal KCNQ potassium channels: physiology and role in
defects caused by missense mutations in S6 segments associated with
disease. Nat Rev Neurosci. 2000;1:21–30.
myotonia: S804F and V1293I. J Physiol. 1998;510:685– 694.
3. Jentsch TJ, Schroeder BC, Kubisch C, et al. Pathophysiology of KCNQ
29. Wang DW, Viswanathan PC, Balser JR, et al. Clinical, genetic, and
channels: neonatal epilepsy and progressive deafness. Epilepsia. 2000;
biophysical characterization of SCN5A mutations associated with atrio-
ventricular conduction block. Circulation. 2002;105:341–346.
EPATITE B: DIAGNOSI. Il Laboratorio GA Niro, A Andriulli Divisione di Gastroenterologia - Casa Sollievo della Sofferenza- IRCCS San La descrizione del meccanismo replicativo del virus dell’epatite B (HBV), nel 1980, el’introduzione del metodo PCR (polymerase chain reaction), nel 1990, hanno rappresentato tappeconoscitive essenziali nel campo virologico. Sono state messe a punto metodi
INTRODUCTION or other important areas of functioning. Contraindications to cosmetic procedures fall(3) It cannot be better explained by anotherinto several categories. Obvious medical issuesmental disorder (eg dissatisfaction in size andsuch as bleeding disorders or allergies soonbody shape in anorexia nervosa, chronic painbecome apparent as the history is taken. Poorsurgical candidates
 Basic Science Reports
Basic Science Reports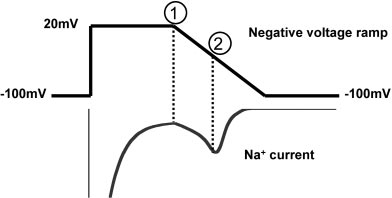 2234
2234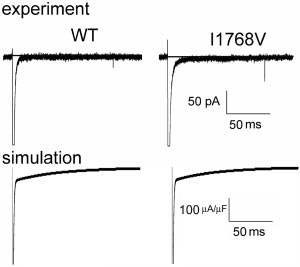
 Clancy et al
Clancy et al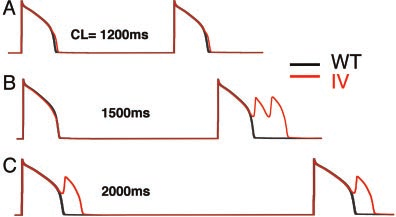
 2236
2236