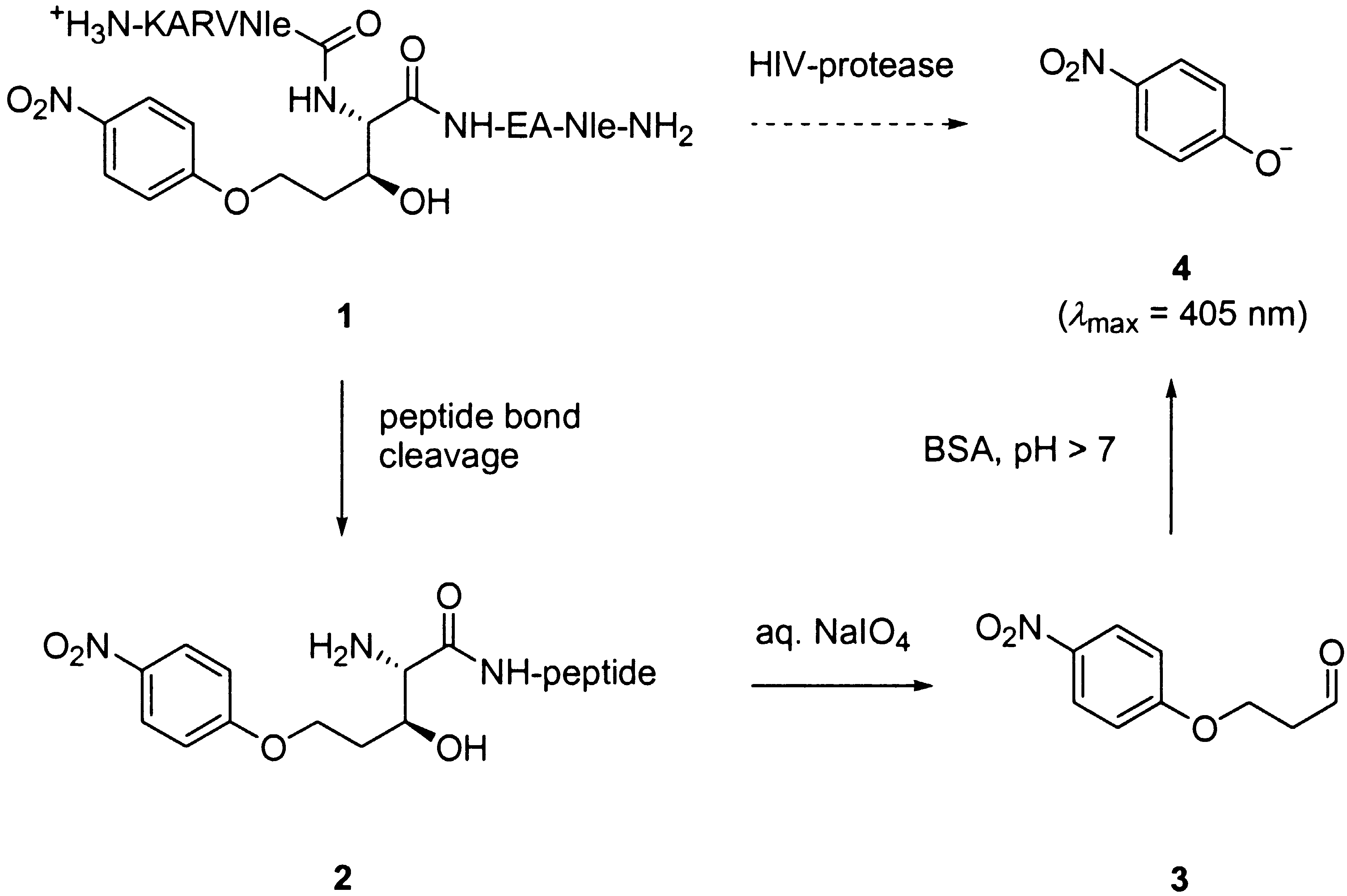
 but the signal modulation produced is only marginal, and, therefore, quite difficult to
observe. Herein, we report a related assay for HIV-protease based on the use of peptide
1 incorporating a non-natural allo-threonine analog at the amino-side of the scissile
peptide bond (Scheme 1). This amino acid undergoes a chromogenic reaction when its
amino-group is free, following an oxidation/b-elimination sequence.
but the signal modulation produced is only marginal, and, therefore, quite difficult to
observe. Herein, we report a related assay for HIV-protease based on the use of peptide
1 incorporating a non-natural allo-threonine analog at the amino-side of the scissile
peptide bond (Scheme 1). This amino acid undergoes a chromogenic reaction when its
amino-group is free, following an oxidation/b-elimination sequence.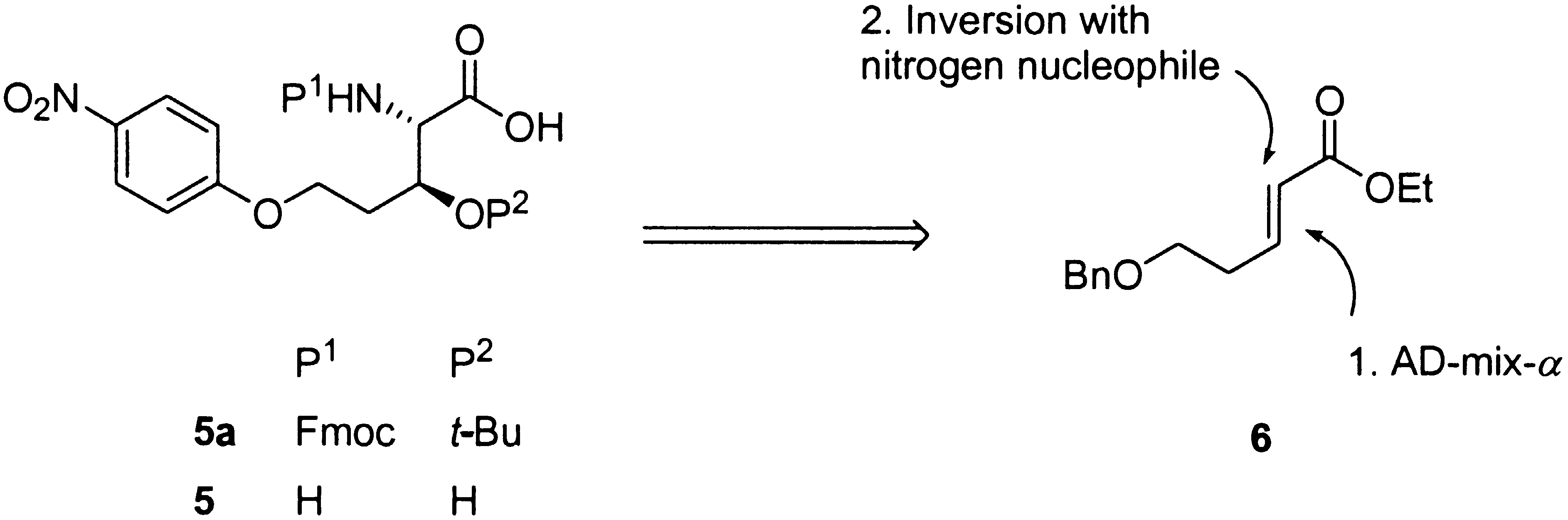 protecting group on the b-OH group would be achieved along the synthesis by standard
Scheme 2. Retrosynthetic Analysis for the Target Amino Acid 5
The synthesis was realized as follows (Scheme 3). Asymmetric dihydroxylation of 6
with AD-mix-a gave diol 7 in 73% and good optical purity (95% ee) [12]. The diol
function was then protected as an acetonide to provide ester 8 (98%). The cyclic acetal
locked the conformation of the molecule and prevented any interaction between the
ester function and the OÀC(5) group, which could then be deprotected by hydro-
genation to give 9 quantitatively without any lactonization. Mitsunobu reaction to the
corresponding iodide 10 (90%) [13] and reaction with sodium 4-nitrophenolate in DMF
gave ether 11 (89%). The acetonide was then removed by acidic treatment in absolute
EtOH to give diol 12 with a disappointing yield of 70% despite many attempts to
optimize. According to a known sequence [14], this diol reacted with 2-nitro-
phenylsulfonyl chloride to give 13 (75%), which was treated with NaN3 to give azido
alcohol 14 (97%). At that stage, the OH group was protected as tert-butyl ether to give
15 via reaction with 2-methylpropene in the presence of H2SO4 (77%). The azido group
was then reduced with PPh3 to give 16. Finally, the ester function was saponified by
treatement with LiOH, and the crude amino acid was converted to the Fmoc-derivative
5a, which was isolated as pure product in 71% yield over the last three steps. The
desired chromogenic amino acid was thus obtained in a total of eleven steps and in 16%
Two chromogenic peptide substrates were prepared by Fmoc-solid-phase peptide
synthesis on Rink-amide polystyrene resin incorporating the protected amino acid 5a.
protecting group on the b-OH group would be achieved along the synthesis by standard
Scheme 2. Retrosynthetic Analysis for the Target Amino Acid 5
The synthesis was realized as follows (Scheme 3). Asymmetric dihydroxylation of 6
with AD-mix-a gave diol 7 in 73% and good optical purity (95% ee) [12]. The diol
function was then protected as an acetonide to provide ester 8 (98%). The cyclic acetal
locked the conformation of the molecule and prevented any interaction between the
ester function and the OÀC(5) group, which could then be deprotected by hydro-
genation to give 9 quantitatively without any lactonization. Mitsunobu reaction to the
corresponding iodide 10 (90%) [13] and reaction with sodium 4-nitrophenolate in DMF
gave ether 11 (89%). The acetonide was then removed by acidic treatment in absolute
EtOH to give diol 12 with a disappointing yield of 70% despite many attempts to
optimize. According to a known sequence [14], this diol reacted with 2-nitro-
phenylsulfonyl chloride to give 13 (75%), which was treated with NaN3 to give azido
alcohol 14 (97%). At that stage, the OH group was protected as tert-butyl ether to give
15 via reaction with 2-methylpropene in the presence of H2SO4 (77%). The azido group
was then reduced with PPh3 to give 16. Finally, the ester function was saponified by
treatement with LiOH, and the crude amino acid was converted to the Fmoc-derivative
5a, which was isolated as pure product in 71% yield over the last three steps. The
desired chromogenic amino acid was thus obtained in a total of eleven steps and in 16%
Two chromogenic peptide substrates were prepared by Fmoc-solid-phase peptide
synthesis on Rink-amide polystyrene resin incorporating the protected amino acid 5a.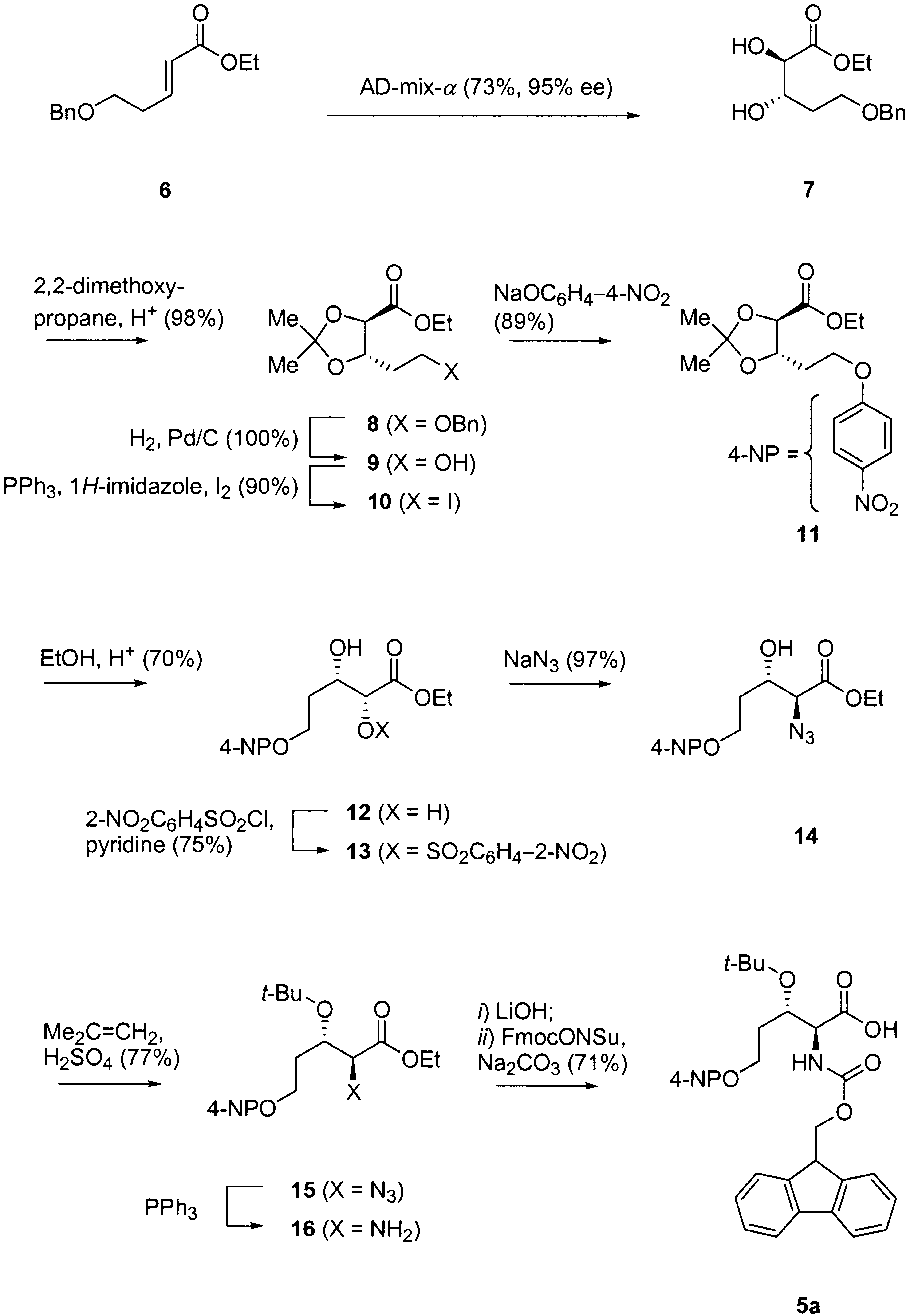 Scheme 3. Stereoselective Synthesis of Chromogenic Amino Acid 5a
amount of 4-nitrophenol produced was then determined spectrophotometrically at
405 nm. The entire assay sequence was conveniently carried out in a total volume of
0.15 ml in individual wells of 96-well microtiter plates.
Scheme 3. Stereoselective Synthesis of Chromogenic Amino Acid 5a
amount of 4-nitrophenol produced was then determined spectrophotometrically at
405 nm. The entire assay sequence was conveniently carried out in a total volume of
0.15 ml in individual wells of 96-well microtiter plates.Lapwing recovery project
Upper Onny Wildlife Group LAPWING RECOVERY PROJECT Together with Shropshire Wildlife Trust Lapwing Project © John Harding © John Harding Case Study & Fact Sheet Practical Farm Management for Breeding Lapwings are in danger of becoming extinct as a breeding bird in many parts of Shropshire, including the Upper Onny area, so the Wildl