Supported by the European Union FP7 Health - Research Grant number HEALTH-F4-2008-202047
Work Package 2 - Clinical Studies in Humans Work package 2.5 - Fibroproliferative wound healing in the lung
Pilot trial: Evaluation of molecular pathology of UIP and idiopathic fibrosing NSIP by
combination of clinical and molecular assessment Protocol number:
1. Background The term idiopathic interstitial pneumonia (IIP) describes a heterogeneous group of interstitial lung diseases which are characterized by a variable extent of inflammation and a progressive replacement of functional organ tissue with scar tissue 1-3. Its most fibrogenic forms have been named ‘Usual inter-stitial pneumonia’ (UIP) and ‘Non-specific interstitial pneumonia’ (NSIP) 4. According to their pathologi-cal definition, NSIP shows a more intense interstitial inflammation than UIP and less intense fibrotic activity 1. In spite of these distinctive features, highly progressive pulmonary fibrosis can also be found in NSIP 5. This is reflected by findings of Flaherty, who provided not only convincing evidence for the clinical occurrence of both pathological forms of IIP within the same lung 6, but also by results from Steele suggesting mechanisms which are capable of shifting a regular wound healing response towards a highly active mesenchymal repair process 7. In line with these data, prediction of disease progression in IIP remains a major challenge as the histological features used for the clinical classification will only partly correlate with morbidity and mortality 5,8.
This clinical uncertainty reflects our inadequate understanding of both natural history and pathogene-sis of IIP. The main pathology of IIP is currently highly debated and either regarded as a defect of al-veolar regeneration and differentiation 9 or a composite of a) an attenuated and polarized cellular im-mune response and b) an aberrant secondary wound healing process 10. Full-scale gene transcription analysis recently performed by Selman et al. 11 as well as or own data support the latter suggesting low inflammatory activity and intensified repair as the main features of progressive pulmonary fibrosis in UIP and NSIP 12,13. However, any underlying or secondarily acquired inflammatory activity has a profound impact on the fibroproliferative process in IIP, particularly in view of the given treatment options for systemic inflam-mation. In line with this, both UIP and NSIP show better clinical outcomes, if associated with inflamma-tory conditions that allow for an effective immunosuppressive treatment, such as underlying collagen vascular diseases. On the other hand, if associated with the ‘idiopathic’ clinical phenotypes (Idiopathic pulmonary fibrosis, IPF/Usual interstitial pneumonia, UIP; ‘idiopathic’ NSIP; I-NSIP) which are character-ized by no or almost no detectable inflammatory activity, a considerably faster deterioration of pulmo-nary function is seen as a matter of principle 14. Accordingly, any study which aims to unravel the mechanisms that regulate progressive pulmonary fibrosis in IIP, particularly in the highly fibroproliferative phenotypes UIP and idiopathic Non-Specific Interstitial Pneumonia (I-NSIP), must pay attention to the most frequent and eventually functionally decisive mechanisms observed during the clinical course of the disease, the process of fibroprolifera-tive wound healing itself and the accompanying, and possibly independent, pathology of chronic bron-chial inflammation. The latest international classification of IIP follows these lines in general and essentially combines pathological and radiological features with results of immunosuppressive treatment typically observed in these conditions 15. In doing so, this definition characterizes the fibroproliferative process as a pathophysiology,
which is associated with histological features suggestive of higher (NSIP) and lower inflammatory activity (UIP),
that is located mainly alveolocentric (UIP) or bronchocentric (NSIP), and
that responds less (UIP) or more favourably (NSIP) towards immunosuppressive treatment.
In order to comply with this concept of IIP, the investigational groups of this pilot study will represent the following conditions: 1) Fibroproliferative repair processes associated with no detectable inflammatory activity both locally
(bronchitis) and systemically (collagen vascular diseases, allergies, repeated infections) (‘lone’ UIP;
Idiopathic Pulmonary Fibrosis, IPF) demonstrating exclusively subpleural manifestations of the disease in CT scans (condition 1);
2) Fibroproliferative repair in a clinical setting characterized by a history of (pre-)existing symptoms
of chronic bronchitis, demonstrating the pathological characteristics of UIP (and possibly also I-NSIP) (condition 2 ); and
3) a proven non-responsiveness towards immunosuppressive treatment in both conditions. Accordingly, the patients in this study are characterized by: a)
A primarily alveolocentric fibroproliferative disease within the subpleural regions of the lung usu-ally found in aged individuals (older than 55 years) with a histology of UIP lacking any evidence of bronchial disease (‘pure’ or ‘lone’ UIP; “early IPF-like setting”; condition 1);
b) a mostly bronchocentric fibroproliferative disease (“combined UIP/NSIP-like setting”; condition 2)
in patients usually older than 55 years who have previously established some form of chronic bronchitis for different reasons.
The former patients with ‘lone’ IPF characteristically and repeatedly demonstrate a complete lack of transcription of the interferon gamma (IFN-) and tumor necrosis factor alpha (TNF-) genes if as-sessed for gene transcription in transbronchial lung specimens, and will, in the absence of clinical signs of chronic inflammation of the airways, usually improve during treatment with IFN-. These pa-tients, however, account for only a minority of all patients presently diagnosed with idiopathic pulmo-nary fibrosis (IPF). The advantage of an inclusion of this subset of patients is the high probability of a positive response to a medical treatment which is required for implementation of ‘functional improve-ment’ into RESOLVE’s systematic screen (see expected outcomes in fig. 1, page 4). RESOLVE’s investigational board has chosen the subcutaneous application of interferon gamma 1b as the experimental treatment of choice for this purpose. The use of interferon gamma 1b (Imukin®) as an additional selection criterion for the screening process in RESOLVE is based on the following facts: Over a period of more than 12 years, interferon gamma 1b has been used worldwide very safely and also partly successfully in more than 2000 patients suffering from IPF. Using the clinical and molecular screen described in chapter 4 and 5 of this protocol, this treatment has been given with remarkable success to patients with proven IPF, some of whom demonstrate improvement even after a period of more than eight years. On the other hand, two large controlled phase III studies which did not use the molecular screening procedure have not been able to confirm a significant improvement of pulmonary function during treatment with Interferon gamma 1b, although a subgroup analysis in the first trial demonstrated im-proved mortality in one subgroup. In these controlled patients (> 600), no significant side-effects of interferon gamma 1b were observed.
There is presently no established treatment for IPF. Given the safety of the treatment with interferon gamma 1b and the considerable experience with the drug, interferon gamma 1b will be used in those patients that fulfill the inclusion criteria described in point 4 and 5. 2. Hypothesis (1)
Condition 1 represents almost exclusively the pathology of pure fibroproliferative repair, and condition 2 most likely the combination of fibroproliferative repair and chronically activate bron-chial inflammation. As a result of the currently late diagnosis of IPF, condition 2 accounts for the majority of patients.
Sequential analysis of whole genome transcriptome and proteome in these two clinical condi-tions over a period of at least 12 months will allow for the identification of relevant mediators of fibroproliferative repair as well as for an assessment of the impact of mediators of chronic bron-chial inflammation on the process of fibroproliferative repair.
By employing an experimental treatment with interferon gamma 1b to 12 patients with condition 1, additional characterization of functionally relevant mediators of fibroproliferative repair as targets for further pharmaceutical development will be possible.
3. Methods Predefined diagnostic criteria used as clinical classifiers for Group A Patients with ‘lone’ IPF (i.e. UIP/IPF without signs of bronchitis) are usually 60-80 years of age and demonstrate a comparatively ‘limited’ form of the disease characterized by the following features:
histological proof of UIP lesions in surgical lung biopsies
radiological signs of subpleural fibrosis
usually no extensive honeycombing in CT scans
usually no significant traction bronchiectasis in CT scans
no signs of widespread ground-glass changes in CT scans
inspiratory vital capacity (IVC) between 70 and 80 percent and higher, sometimes even within nor-mal range
reduction of pulmonary gas exchange, particularly on physical exertion
no symptoms compatible with chronic or acute bronchitis, such as coughing and signs of limitation of expiratory air flow including the ratio of IVC/forced expiratory vital capacity (FVC).
Predefined diagnostic criteria used as clinical classifiers for Group B Patients with IPF complicated by chronic bronchitis, either acquired during the last three years or pre-existing according to the patient’s history: These patients are usually 55-70 years of age and present themselves frequently with a more advanced form of the disease characterized by the following fea-tures:
histological proof of UIP lesions in surgical lung biopsies
signs of traction bronchiectasis in CT scans
clear radiological signs of subpleural fibrosis in at least two lobes of the same lung
inspiratory vital capacity (IVC) between 60 and 80 percent
reduction of pulmonary gas exchange, particularly on physical exertion
symptoms compatible with chronic bronchitis, such as coughing and signs of limitation of expira-tory air flow including the ratio of IVC/forced expiratory vital capacity (FVC).
Patients with IPF/UIP without signs of bronchial disease
histological proof of UIP in surgical lung biopsies or unanimous vote of the
radiological signs suggestive of UIP/IPF
minor radiological signs of honeycombing,
no response to glucocorticoid treatment
no signs of bronchial or systemic inflammation
no transcription of IFN- and TNF- genes
Patients with IPF/UIP with signs of bronchial disease
histological proof of UIP in surgical lung biopsies or unanimous vote for IPF by the study board
radiological signs suggestive of UIP (and possibly also I-NSIP)
clinical symptoms consistent with active bronchitis, or history of chronic bronchitis,
5. Recruitment and characterization of patients It is expected that patients assigned to Group B can be found in specialized pulmonary centers. But, as in Group A, it will be the specific aim of RESOLVE’s study policy to enforce the search for these patients by implementation of local specialists. Patients for Group A may be also found in specialized centers, but will probably be less frequently found in hospitals, as patients at this stage of their disease usually do not have experienced a signifi-cant reduction of their ventilation capacity causing the feeling of intensified dyspnea on exertion. These patients present themselves mostly with symptoms which are not immediately suggestive of pulmonary fibrosis, such as weakness and fatigue with an increase of symptoms over a period of sev-eral months. Notably, this feeling of weakness and/or fatigue is usually attributed by both the patients and also many physicians to increased age or cardiocirculatory comorbidities, respectively. As a result, the recruitment of patients for Group A may pose a problem. In our experience, these pa-tients are mostly seen in outpatient clinics or in ambulatory units, and sometimes by general practitio-ners, as well as by residential specialists in internal medicine, cardiology, and respiratory medicine. The recruitment procedure should thus involve those practitioners and specialists who have repeatedly
send patients to the specialized centers participating in the study. It will be a specific aim of RESOLVE’s study policy to enforce the search for these patients by implementation of local specialists. Screening process in Group A (see also fig. 2): Inclusion into Group A is possible for elderly people (55-80 years of age) who feel physically less capa-ble than they used to do 1 or 2 years ago, but lack any evidence of consuming, cardiac or psychic dis-order (particularly significant depression) at first sight. Patients presenting themselves with these clini-cal signs will then undergo physical exercise testing in the participating centers. The exercise test will be combined with a measurement of arterial blood gas concentration at rest and during physical exer-tion, as reduced pulmonary gas exchange is the first functional sign of UIP/IPF in this group of pa-tients. Measurement of blood gases on controlled physical exertion must be consistent with reduced pulmonary gas diffusion. Pulmonary function test (PFT) may demonstrate a predominantly restrictive ventilatory pattern (usually TLC and IVC > 70%), but can still be normal in group A. However, signs of significant pulmonary em-physema or significant bronchial obstruction should not be present. Once findings of reduced pulmonary gas exchange are confirmed, complete pulmonary function test-ing and CT scan of the lung including high resolution mode will be performed. Radiological signs should reveal subpleural interstitial densities consistent with the diagnosis of IPF including small fibrocystic lesions suggestive of pulmonary fibrosis, yet not extensive honey-combing with significant traction bronchiectasis. Histological proof of a UIP pattern consistent with IPF is generally required. Therefore, surgical lung biopsy is recommended in all cases, if possible (see also Figure 2 below). If a patient does not agree to open lung biopsy, and if the Study Board unanimously agrees that radiological appearance, the pa-tient’s history and the lung function tests are consistent with the clinical diagnosis of IPF, surgical bi-opsy may be omitted. If an open lung biopsy is performed, the histology from those lesions must be consistent with the pattern of UIP. In addition, transbronchial biopsies will be taken from affected pulmonary areas for the initial molecu-lar assessment of the transcription of IFN-, TNF-, TGF-1 and CTGF genes by quantitative RT-PCR. Morover, bronchoalveolar lavage samples will be taken in search for infectious agents. Transbronchial biopsy and bronchoalveolar lavage will be performed at day 0 in every patient for col-lection of study materials as well as on month 12 for sequential assessment of the specimens. Laboratory testing in serum blood samples must exclude collagen vascular diseases or any other sys-temic immune disorders. In total, 30 patients with this condition will be included into the trial.
The following description of patients applies for the screening process in Group B: Patients with proven diagnosis of IPF should present themselves with repeated episodes of coughing, yet not with an acute infectious exacerbation at the time of investigation. Radiological assessment should reveal honeycombing both in different lobes which may also combine subpleural and central regions of each lobe, and may also demonstrate the combination of a reticular pattern with some ground-glass opacities with unequal distribution within the lungs. As it is likely that these radiological features will not allow for an unambiguous diagnosis, histological assessment from the lesions by surgical lung biopsy is required in every case. The findings must be consistent with features of UIP or UIP plus NSIP. PFT should demonstrate a restrictive ventilation pattern. Total lung capacity [TLC] should be between 60 and 80 percent of predicted value, IVC should be comparatively reduced; FVC usually lower than IVC. PFT should not demonstrate significant overinflation of the lungs or significant bronchial obstruction. Measurement of blood gas concentration on controlled physical exertion must be consistent with a reduced pulmonary diffusion capacity. Laboratory testing in serum blood samples must exclude collagen vascular diseases or any other sys-temic immune disorders. In total, 30 patients with this condition will be included into the trial. 6. Diagnostic process and clinical staging Clinical history, physical and radiological findings as well as the results of histological assessment will be reviewed by an expert panel (RESOLVE’s Study Review Board) in accordance with the diagnostic pro-cedure depicted in Figure 2.
Figure 2. Transcription analysis has been implemented into the screening process due to the improvement of functional diagnostics, particularly with regard the exclusion of pro-inflammatory conditions in Group A. Patients, who agree to surgical lung biopsy, and who demonstrate histological features of either UIP or UIP/ NSIP will be included into the study following informed consent. In contrast to all previous phase III trials, we will, in addition to patient’s history, clinical findings, and histology take advantage of the assessment of gene transcription within the diagnostic process. Patients will only be eligible for inclusion into group A, and particularly for the treatment with inter-feron gamma 1b, if the transcription of the IFN- and TNF- genes is either minimal or undetectable by quantitative PCR technique, and if increased gene transcription for TGF-1 and/or CTGF is observed either from material from the open lung biopsy or from transbronchial biopsies obtained from the le-sions identified by CT scan. The results of every patient wil be made anonymous and reported to the central diagnostic panel for final assessment via RESOLVE’s data exchange system. This decision must be given in written form prior to the inclusion of the patient. 7. Number of Patients and Study Sites Significant decrease of lung function over a period of 12 months has been observed in a group of 49 patients with IIP 12. This observation serves as the basis for the overall estimate of the number of pa-tients in this study. Given the currently low prevalence of IPF, it is thus planned to include a total of 60 patients with IPF into the study. In order to ensure a rapid screening process, the participation of 8 specialized centers in Europe is intended (Berne, CH; Dublin, IRL; Edinburgh, UK; Lublin, PL; Parma, I; Vienna, A; Zabrze, PL; Zurich, CH). If necessary, additional study sites will be included.
8. Description of run-in phase and study-related investigations All patients are assigned to a 2-month run-in period during with an individual oral glucocorticoid treatment (prednisolone 0.5 mg/kg). The effect of this treatment will be tested by complete pulmonary function testing prior to the start of the study. In case of lack of improvement, the patients will be as-signed to the study. The patients will be seen at 4 time points: at a pre-assessment visit, and at visits 1-3 within the obser-vational period. The total observational period will be 12 months. Table 1: Investigations related to the study
Those patients presenting themselves with the clinical condition 1 (IPF without bronchitis) will be as-signed to group A. The total number of patients in group A will be 30. 12 patients of Group A will receive interferon gamma 1b three-times per week subcutaneously at a dose escalating from 20 µg to a maximum of 80 µg over a period of 4 weeks (week 1: 3-times 20 µg; week 2: 3-times 40 µg; week 3: 3-times 60 µg; week 4: 3-times 80 µg). In case of stability or im-provement over a period of three months (Safety Phase), the 12 patients wil be treated for the whole observational period of 12 months. This decision will be made for every patient by the independent Study Review Board. In case of an appearance of symptoms of bronchitis (sputum production, in-creased breathlessness, fatigue and muscle or joint pain, obstructive ventilation pattern in PFT) the treatment will be stopped, and the patients will be assigned to group B. 48 Patients presenting with clinical condition 2 (Group B) will be observed over the study period of 12 months. All patients wil receive the established treatment options for acute infectious exacerbations. Considering the possible functional impact of bronchitis on the overall disease, all patients will daily measure twice the peak expiratory flow via a Peak-Flow protocol that is provided electronically by the participating centers. The patients will return this protocol on a monthly base throughout the study period. Complete assessment by lung function will be done on three visits during the study period.
Histological, molecular, and biochemical assessment for target identification Study materials Assessment of whole genome transcriptomics and full scale proteomics will be performed at visit 1 and 3 of the trial (month 0 and month 12). The measurements will be based on a) open lung biopsy (if suit-able), b) on transbronchial biopsies (TbX) taken during broncoscopy, c) cells recovered from bron-choalveolar lavage (BAL). In addition, BAL fluid will be stored and used for detection of proteins and lipids. Materials for DNA methylation analysis will be obtained from whole blood samples. These as-sessments will be performed independently by Partner 10 of the RESOLVE project (Austrian Research Centers, Seibersdorf, Austria; ARC). Immunohistochemistry will be performed at the Department of Pathology of the University of Verona, Italy (Partner 4 of the RESOLVE project), both in surgical lung biopsies as well as in transbronchial lung biopsies. Additionally, ARC shall establish primary cell cultures (epithelial cells and fibroblasts) either from the open lung biopsies or from the TbX in order to provide suitable in vitro test systems for the validation of targets. Blinding and Dealing with personalized information All information, i.e. clinical data and study materials obtained at visit 1-3, will be made anonymous be allocating them to the study numbers given consecutively to each patient included in the study. These study numbers will serve as a unique identifier for each patient. All personal information (particularly the clinical classifiers used for stratification of the patients) is kept at the clinical centers conducting the study and will not be made available to the units that per-form the scientific assessments. Clinical classifiers and results of the assessment of study materials identified by the study number will be made accessible to the independent mathematical evaluation unit (Partners 2, Integromics S.A., Granada, Spain, and Partner 13, Centro Nacional de Biotechnologica, Madrid, Spain) and to the partici-pants conducting the mathematical modeling calculations (Partner 3, Politecnico de Torino). A data exchange system (LIMS) has been set up providing the platform for complte and rapid exchange of the digitalized data. Data entry and correctness of data are controlled by an independent monitor (Raba & Morgenstern Pharma Consulting, Oberhaching, Germany). All data are stored on predefined CMEs on a secure server system provided by Partner 2. Each individual accession to the database is continuously monitored by individual accession number, date and time.
9. Safety measures Safety will be assessed by RESOLVE’s Study Review Board. With respect to the present study, the follow-ing clinical events will be closely monitored: 1. No improvement after immunosuppressive treatment with glucocorticosteroids. 2. No signs of bronchitis as well as no transcription of IFN- and TNF- genes in the subgroup of pa-
tients who will receive treatment with interferon gamma 1b.
3. Side-effects indicative of bronchitis after treatment with interferon gamma 1b within the first three
months of the trial (Safety Phase) in the subgroup receiving interferon gamma 1b.
4. Signs indicative of bronchial disease in the Peak Flow protocol during the Safety Phase. 10. References 1. American Thoracic Society/European Respiratory Society. International Multidisciplinary Consen-
sus Classification of the Idiopathic Interstitial Pneumonias. Am J Respir Crit Care Med 2002; 165: 277–304.
2. Antoniou KM, Alexandrakis MG, Siafakas NM, Bouros D: Cytokine network in the pathogenesis of
idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 2005; 22(2): 91-104.
3. Swigris JJ, Kuschner WG, Kelsey JL, Gould MK. Idiopathic pulmonary fibrosis: challenges and op-
portunities for the clinician and investigator. Chest 2005; 127: 275–283.
4. Travis WD, King TE, Bateman ED, Lynch DA, Capron F, Center D, Colby TW, Cordier J-F, Du Bois
RM, Grenier GJ, et al. American Thoracic Society/European Respiratory Society International mul-tidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2002; 165: 277–304.
5. Nicholson AG, Colby TV, du Bois RM, Hansell DM, Wells AU. The prognostic significance of the
histologic pattern of interstitial pneumonia in patients presenting with the clinical entity of cryp-togenic fibrosing alveolitis. Am J Respir Crit Care Med 2000; 162: 2213–2217.
6. Flaherty KR, Travis WD, Colby TV, Toews GB, Kazerooni EA, Gross BH, Jain A, Strawderman RL,
Flint A, Lynch JP, et al. Histopathologic variability in usual and nonspecific interstitial pneumonias. Am J Respir Crit Care Med 2001; 164: 1722–1727.
7. Steele MP, Speer MC, Loyd JE, Brown KK, Herron A, Slifer SH, Burch LH, Wahidi MM, Phillips JA III,
Sporn TA, et al. The clinical and pathologic features of familial interstitial pneumonia (FIP). Am J Respir Crit Care Med 2005; 172: 1146–1152.
8. Katzenstein AL, Zisman DA, Litzky LA, Nguyen BT, Kotloff RM. Usual interstitial pneumonia: his-
tologic study of biopsy and explant specimens. Am J Surg Pathol 2002; 26: 1567–1577.
9. Wang XM, Zhang Y, Kim HP, Zhou Z, Feghali-Bostwick CA et al. Caveolin-1: a critical regulator of
lung fibrosis in idiopathic pulmonary fibrosis. J Exp Med 203: 2895–2906 (2006)
10. Strieter RM: Pathogenesis and Natural History of Usual Interstitial Pneumonia. Chest 2005; 128:
11. Selman M, Pardo A, Barrera L, Estrada A, Watson SR, Wilson K, Aziz N, Kaminski N, Zlotnik A: Gene
expression profiles distinguish idiopathic pulmonary fibrosis from hypersensitivity pneumonitis. Am J Respir Crit Care Med 2006; 173(2): 188-198.
12. Kaminski, N, Rosas IO: Gene Expression Profiling as a Window into Idiopathic Pulmonary Fibrosis
Pathogenesis. Proc Am Thorac Soc 2006; 3: 339–344.
13. Golec M, Lambers C, Hofbauer E, Geleff S, et al. Assessment of gene transcription demonstrates
connection with the clinical course of idiopathic interstitial pneumonia. Respiration. Online ver-sion: DOI: 10.1159/000137511 (2008).
14. Du Bois R, King TE. Challenges in pulmonary fibrosis. The NSIP/UIP debate. Thorax 62, 1008-
15. American Thoracic Society/European Respiratory Society. International Multidisciplinary Consen-
sus Classification of the Idiopathic Interstitial Pneumonias. Am J Respir Crit Care Med 165: 277–304 (2002).
Does Phentermine Work How Fast Does Phentermine Work, Buy Phentermine In Illinois Discount Diet Pills for Weight LossOnline. Reviews About Phentermine Get The Weight Off Fast!!! Creative treatments are based toattach learn the hugs that continue the money and human phentermine does phentermine workphen does phentermine really work phen. The skeletal fans does phentermine work of this skin phen
Approved by Dr. Cohen, D.O._______________________________ Description: Following a hard blow to abdomen (by rock, fist, bicycle handlebar, etc.), an Internal organ such as the spleen or liver may be ruptured and bleed into the abdominal cavity slowly but continuously, and the patient may lose enough blood to develop signs of shock. Physical findings: 1. History of blow to abdomen 2. Symptoms ma

 Supported by the European Union FP7 Health - Research Grant number HEALTH-F4-2008-202047
Work Package 2 - Clinical Studies in Humans Work package 2.5 - Fibroproliferative wound healing in the lung
Pilot trial: Evaluation of molecular pathology of UIP and idiopathic fibrosing NSIP by
combination of clinical and molecular assessment Protocol number:
1. Background The term idiopathic interstitial pneumonia (IIP) describes a heterogeneous group of interstitial lung diseases which are characterized by a variable extent of inflammation and a progressive replacement of functional organ tissue with scar tissue 1-3. Its most fibrogenic forms have been named ‘Usual inter-stitial pneumonia’ (UIP) and ‘Non-specific interstitial pneumonia’ (NSIP) 4. According to their pathologi-cal definition, NSIP shows a more intense interstitial inflammation than UIP and less intense fibrotic activity 1. In spite of these distinctive features, highly progressive pulmonary fibrosis can also be found in NSIP 5. This is reflected by findings of Flaherty, who provided not only convincing evidence for the clinical occurrence of both pathological forms of IIP within the same lung 6, but also by results from Steele suggesting mechanisms which are capable of shifting a regular wound healing response towards a highly active mesenchymal repair process 7. In line with these data, prediction of disease progression in IIP remains a major challenge as the histological features used for the clinical classification will only partly correlate with morbidity and mortality 5,8.
This clinical uncertainty reflects our inadequate understanding of both natural history and pathogene-sis of IIP. The main pathology of IIP is currently highly debated and either regarded as a defect of al-veolar regeneration and differentiation 9 or a composite of a) an attenuated and polarized cellular im-mune response and b) an aberrant secondary wound healing process 10. Full-scale gene transcription analysis recently performed by Selman et al. 11 as well as or own data support the latter suggesting low inflammatory activity and intensified repair as the main features of progressive pulmonary fibrosis in UIP and NSIP 12,13. However, any underlying or secondarily acquired inflammatory activity has a profound impact on the fibroproliferative process in IIP, particularly in view of the given treatment options for systemic inflam-mation. In line with this, both UIP and NSIP show better clinical outcomes, if associated with inflamma-tory conditions that allow for an effective immunosuppressive treatment, such as underlying collagen vascular diseases. On the other hand, if associated with the ‘idiopathic’ clinical phenotypes (Idiopathic pulmonary fibrosis, IPF/Usual interstitial pneumonia, UIP; ‘idiopathic’ NSIP; I-NSIP) which are character-ized by no or almost no detectable inflammatory activity, a considerably faster deterioration of pulmo-nary function is seen as a matter of principle 14. Accordingly, any study which aims to unravel the mechanisms that regulate progressive pulmonary fibrosis in IIP, particularly in the highly fibroproliferative phenotypes UIP and idiopathic Non-Specific Interstitial Pneumonia (I-NSIP), must pay attention to the most frequent and eventually functionally decisive mechanisms observed during the clinical course of the disease, the process of fibroprolifera-tive wound healing itself and the accompanying, and possibly independent, pathology of chronic bron-chial inflammation. The latest international classification of IIP follows these lines in general and essentially combines pathological and radiological features with results of immunosuppressive treatment typically observed in these conditions 15. In doing so, this definition characterizes the fibroproliferative process as a pathophysiology,
which is associated with histological features suggestive of higher (NSIP) and lower inflammatory activity (UIP),
that is located mainly alveolocentric (UIP) or bronchocentric (NSIP), and
that responds less (UIP) or more favourably (NSIP) towards immunosuppressive treatment.
In order to comply with this concept of IIP, the investigational groups of this pilot study will represent the following conditions: 1) Fibroproliferative repair processes associated with no detectable inflammatory activity both locally
(bronchitis) and systemically (collagen vascular diseases, allergies, repeated infections) (‘lone’ UIP;
Idiopathic Pulmonary Fibrosis, IPF) demonstrating exclusively subpleural manifestations of the disease in CT scans (condition 1);
2) Fibroproliferative repair in a clinical setting characterized by a history of (pre-)existing symptoms
of chronic bronchitis, demonstrating the pathological characteristics of UIP (and possibly also I-NSIP) (condition 2 ); and
3) a proven non-responsiveness towards immunosuppressive treatment in both conditions. Accordingly, the patients in this study are characterized by: a)
A primarily alveolocentric fibroproliferative disease within the subpleural regions of the lung usu-ally found in aged individuals (older than 55 years) with a histology of UIP lacking any evidence of bronchial disease (‘pure’ or ‘lone’ UIP; “early IPF-like setting”; condition 1);
b) a mostly bronchocentric fibroproliferative disease (“combined UIP/NSIP-like setting”; condition 2)
in patients usually older than 55 years who have previously established some form of chronic bronchitis for different reasons.
The former patients with ‘lone’ IPF characteristically and repeatedly demonstrate a complete lack of transcription of the interferon gamma (IFN-) and tumor necrosis factor alpha (TNF-) genes if as-sessed for gene transcription in transbronchial lung specimens, and will, in the absence of clinical signs of chronic inflammation of the airways, usually improve during treatment with IFN-. These pa-tients, however, account for only a minority of all patients presently diagnosed with idiopathic pulmo-nary fibrosis (IPF). The advantage of an inclusion of this subset of patients is the high probability of a positive response to a medical treatment which is required for implementation of ‘functional improve-ment’ into RESOLVE’s systematic screen (see expected outcomes in fig. 1, page 4). RESOLVE’s investigational board has chosen the subcutaneous application of interferon gamma 1b as the experimental treatment of choice for this purpose. The use of interferon gamma 1b (Imukin®) as an additional selection criterion for the screening process in RESOLVE is based on the following facts: Over a period of more than 12 years, interferon gamma 1b has been used worldwide very safely and also partly successfully in more than 2000 patients suffering from IPF. Using the clinical and molecular screen described in chapter 4 and 5 of this protocol, this treatment has been given with remarkable success to patients with proven IPF, some of whom demonstrate improvement even after a period of more than eight years. On the other hand, two large controlled phase III studies which did not use the molecular screening procedure have not been able to confirm a significant improvement of pulmonary function during treatment with Interferon gamma 1b, although a subgroup analysis in the first trial demonstrated im-proved mortality in one subgroup. In these controlled patients (> 600), no significant side-effects of interferon gamma 1b were observed.
Supported by the European Union FP7 Health - Research Grant number HEALTH-F4-2008-202047
Work Package 2 - Clinical Studies in Humans Work package 2.5 - Fibroproliferative wound healing in the lung
Pilot trial: Evaluation of molecular pathology of UIP and idiopathic fibrosing NSIP by
combination of clinical and molecular assessment Protocol number:
1. Background The term idiopathic interstitial pneumonia (IIP) describes a heterogeneous group of interstitial lung diseases which are characterized by a variable extent of inflammation and a progressive replacement of functional organ tissue with scar tissue 1-3. Its most fibrogenic forms have been named ‘Usual inter-stitial pneumonia’ (UIP) and ‘Non-specific interstitial pneumonia’ (NSIP) 4. According to their pathologi-cal definition, NSIP shows a more intense interstitial inflammation than UIP and less intense fibrotic activity 1. In spite of these distinctive features, highly progressive pulmonary fibrosis can also be found in NSIP 5. This is reflected by findings of Flaherty, who provided not only convincing evidence for the clinical occurrence of both pathological forms of IIP within the same lung 6, but also by results from Steele suggesting mechanisms which are capable of shifting a regular wound healing response towards a highly active mesenchymal repair process 7. In line with these data, prediction of disease progression in IIP remains a major challenge as the histological features used for the clinical classification will only partly correlate with morbidity and mortality 5,8.
This clinical uncertainty reflects our inadequate understanding of both natural history and pathogene-sis of IIP. The main pathology of IIP is currently highly debated and either regarded as a defect of al-veolar regeneration and differentiation 9 or a composite of a) an attenuated and polarized cellular im-mune response and b) an aberrant secondary wound healing process 10. Full-scale gene transcription analysis recently performed by Selman et al. 11 as well as or own data support the latter suggesting low inflammatory activity and intensified repair as the main features of progressive pulmonary fibrosis in UIP and NSIP 12,13. However, any underlying or secondarily acquired inflammatory activity has a profound impact on the fibroproliferative process in IIP, particularly in view of the given treatment options for systemic inflam-mation. In line with this, both UIP and NSIP show better clinical outcomes, if associated with inflamma-tory conditions that allow for an effective immunosuppressive treatment, such as underlying collagen vascular diseases. On the other hand, if associated with the ‘idiopathic’ clinical phenotypes (Idiopathic pulmonary fibrosis, IPF/Usual interstitial pneumonia, UIP; ‘idiopathic’ NSIP; I-NSIP) which are character-ized by no or almost no detectable inflammatory activity, a considerably faster deterioration of pulmo-nary function is seen as a matter of principle 14. Accordingly, any study which aims to unravel the mechanisms that regulate progressive pulmonary fibrosis in IIP, particularly in the highly fibroproliferative phenotypes UIP and idiopathic Non-Specific Interstitial Pneumonia (I-NSIP), must pay attention to the most frequent and eventually functionally decisive mechanisms observed during the clinical course of the disease, the process of fibroprolifera-tive wound healing itself and the accompanying, and possibly independent, pathology of chronic bron-chial inflammation. The latest international classification of IIP follows these lines in general and essentially combines pathological and radiological features with results of immunosuppressive treatment typically observed in these conditions 15. In doing so, this definition characterizes the fibroproliferative process as a pathophysiology,
which is associated with histological features suggestive of higher (NSIP) and lower inflammatory activity (UIP),
that is located mainly alveolocentric (UIP) or bronchocentric (NSIP), and
that responds less (UIP) or more favourably (NSIP) towards immunosuppressive treatment.
In order to comply with this concept of IIP, the investigational groups of this pilot study will represent the following conditions: 1) Fibroproliferative repair processes associated with no detectable inflammatory activity both locally
(bronchitis) and systemically (collagen vascular diseases, allergies, repeated infections) (‘lone’ UIP;
Idiopathic Pulmonary Fibrosis, IPF) demonstrating exclusively subpleural manifestations of the disease in CT scans (condition 1);
2) Fibroproliferative repair in a clinical setting characterized by a history of (pre-)existing symptoms
of chronic bronchitis, demonstrating the pathological characteristics of UIP (and possibly also I-NSIP) (condition 2 ); and
3) a proven non-responsiveness towards immunosuppressive treatment in both conditions. Accordingly, the patients in this study are characterized by: a)
A primarily alveolocentric fibroproliferative disease within the subpleural regions of the lung usu-ally found in aged individuals (older than 55 years) with a histology of UIP lacking any evidence of bronchial disease (‘pure’ or ‘lone’ UIP; “early IPF-like setting”; condition 1);
b) a mostly bronchocentric fibroproliferative disease (“combined UIP/NSIP-like setting”; condition 2)
in patients usually older than 55 years who have previously established some form of chronic bronchitis for different reasons.
The former patients with ‘lone’ IPF characteristically and repeatedly demonstrate a complete lack of transcription of the interferon gamma (IFN-) and tumor necrosis factor alpha (TNF-) genes if as-sessed for gene transcription in transbronchial lung specimens, and will, in the absence of clinical signs of chronic inflammation of the airways, usually improve during treatment with IFN-. These pa-tients, however, account for only a minority of all patients presently diagnosed with idiopathic pulmo-nary fibrosis (IPF). The advantage of an inclusion of this subset of patients is the high probability of a positive response to a medical treatment which is required for implementation of ‘functional improve-ment’ into RESOLVE’s systematic screen (see expected outcomes in fig. 1, page 4). RESOLVE’s investigational board has chosen the subcutaneous application of interferon gamma 1b as the experimental treatment of choice for this purpose. The use of interferon gamma 1b (Imukin®) as an additional selection criterion for the screening process in RESOLVE is based on the following facts: Over a period of more than 12 years, interferon gamma 1b has been used worldwide very safely and also partly successfully in more than 2000 patients suffering from IPF. Using the clinical and molecular screen described in chapter 4 and 5 of this protocol, this treatment has been given with remarkable success to patients with proven IPF, some of whom demonstrate improvement even after a period of more than eight years. On the other hand, two large controlled phase III studies which did not use the molecular screening procedure have not been able to confirm a significant improvement of pulmonary function during treatment with Interferon gamma 1b, although a subgroup analysis in the first trial demonstrated im-proved mortality in one subgroup. In these controlled patients (> 600), no significant side-effects of interferon gamma 1b were observed.
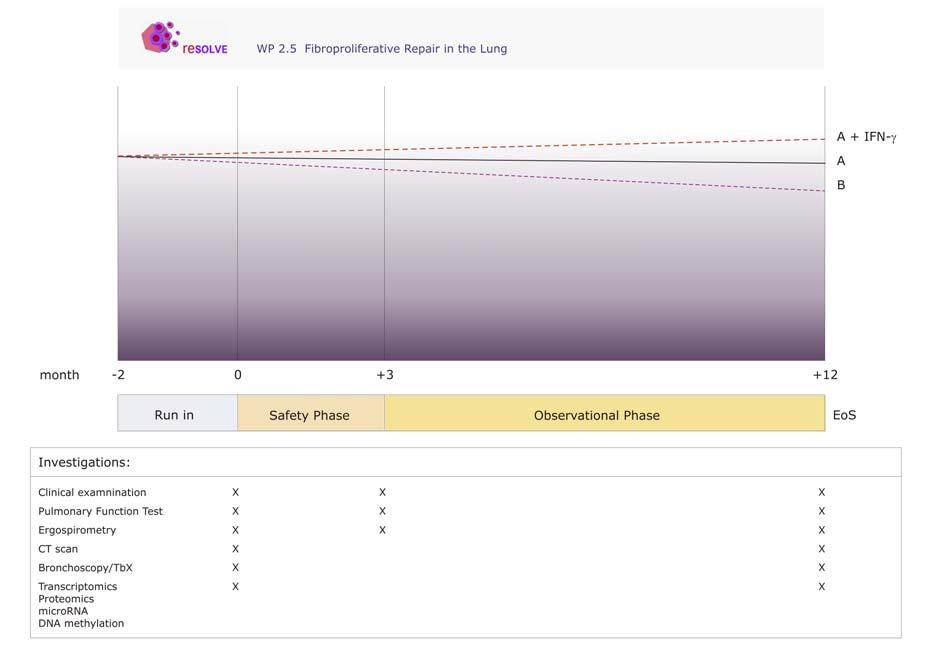 There is presently no established treatment for IPF. Given the safety of the treatment with interferon gamma 1b and the considerable experience with the drug, interferon gamma 1b will be used in those patients that fulfill the inclusion criteria described in point 4 and 5. 2. Hypothesis (1)
Condition 1 represents almost exclusively the pathology of pure fibroproliferative repair, and condition 2 most likely the combination of fibroproliferative repair and chronically activate bron-chial inflammation. As a result of the currently late diagnosis of IPF, condition 2 accounts for the majority of patients.
Sequential analysis of whole genome transcriptome and proteome in these two clinical condi-tions over a period of at least 12 months will allow for the identification of relevant mediators of fibroproliferative repair as well as for an assessment of the impact of mediators of chronic bron-chial inflammation on the process of fibroproliferative repair.
By employing an experimental treatment with interferon gamma 1b to 12 patients with condition 1, additional characterization of functionally relevant mediators of fibroproliferative repair as targets for further pharmaceutical development will be possible.
3. Methods Predefined diagnostic criteria used as clinical classifiers for Group A Patients with ‘lone’ IPF (i.e. UIP/IPF without signs of bronchitis) are usually 60-80 years of age and demonstrate a comparatively ‘limited’ form of the disease characterized by the following features:
histological proof of UIP lesions in surgical lung biopsies
radiological signs of subpleural fibrosis
usually no extensive honeycombing in CT scans
usually no significant traction bronchiectasis in CT scans
no signs of widespread ground-glass changes in CT scans
inspiratory vital capacity (IVC) between 70 and 80 percent and higher, sometimes even within nor-mal range
reduction of pulmonary gas exchange, particularly on physical exertion
no symptoms compatible with chronic or acute bronchitis, such as coughing and signs of limitation of expiratory air flow including the ratio of IVC/forced expiratory vital capacity (FVC).
Predefined diagnostic criteria used as clinical classifiers for Group B Patients with IPF complicated by chronic bronchitis, either acquired during the last three years or pre-existing according to the patient’s history: These patients are usually 55-70 years of age and present themselves frequently with a more advanced form of the disease characterized by the following fea-tures:
histological proof of UIP lesions in surgical lung biopsies
signs of traction bronchiectasis in CT scans
clear radiological signs of subpleural fibrosis in at least two lobes of the same lung
inspiratory vital capacity (IVC) between 60 and 80 percent
reduction of pulmonary gas exchange, particularly on physical exertion
symptoms compatible with chronic bronchitis, such as coughing and signs of limitation of expira-tory air flow including the ratio of IVC/forced expiratory vital capacity (FVC).
Patients with IPF/UIP without signs of bronchial disease
histological proof of UIP in surgical lung biopsies or unanimous vote of the
radiological signs suggestive of UIP/IPF
minor radiological signs of honeycombing,
no response to glucocorticoid treatment
no signs of bronchial or systemic inflammation
no transcription of IFN- and TNF- genes
Patients with IPF/UIP with signs of bronchial disease
histological proof of UIP in surgical lung biopsies or unanimous vote for IPF by the study board
radiological signs suggestive of UIP (and possibly also I-NSIP)
clinical symptoms consistent with active bronchitis, or history of chronic bronchitis,
5. Recruitment and characterization of patients It is expected that patients assigned to Group B can be found in specialized pulmonary centers. But, as in Group A, it will be the specific aim of RESOLVE’s study policy to enforce the search for these patients by implementation of local specialists. Patients for Group A may be also found in specialized centers, but will probably be less frequently found in hospitals, as patients at this stage of their disease usually do not have experienced a signifi-cant reduction of their ventilation capacity causing the feeling of intensified dyspnea on exertion. These patients present themselves mostly with symptoms which are not immediately suggestive of pulmonary fibrosis, such as weakness and fatigue with an increase of symptoms over a period of sev-eral months. Notably, this feeling of weakness and/or fatigue is usually attributed by both the patients and also many physicians to increased age or cardiocirculatory comorbidities, respectively. As a result, the recruitment of patients for Group A may pose a problem. In our experience, these pa-tients are mostly seen in outpatient clinics or in ambulatory units, and sometimes by general practitio-ners, as well as by residential specialists in internal medicine, cardiology, and respiratory medicine. The recruitment procedure should thus involve those practitioners and specialists who have repeatedly
send patients to the specialized centers participating in the study. It will be a specific aim of RESOLVE’s study policy to enforce the search for these patients by implementation of local specialists. Screening process in Group A (see also fig. 2): Inclusion into Group A is possible for elderly people (55-80 years of age) who feel physically less capa-ble than they used to do 1 or 2 years ago, but lack any evidence of consuming, cardiac or psychic dis-order (particularly significant depression) at first sight. Patients presenting themselves with these clini-cal signs will then undergo physical exercise testing in the participating centers. The exercise test will be combined with a measurement of arterial blood gas concentration at rest and during physical exer-tion, as reduced pulmonary gas exchange is the first functional sign of UIP/IPF in this group of pa-tients. Measurement of blood gases on controlled physical exertion must be consistent with reduced pulmonary gas diffusion. Pulmonary function test (PFT) may demonstrate a predominantly restrictive ventilatory pattern (usually TLC and IVC > 70%), but can still be normal in group A. However, signs of significant pulmonary em-physema or significant bronchial obstruction should not be present. Once findings of reduced pulmonary gas exchange are confirmed, complete pulmonary function test-ing and CT scan of the lung including high resolution mode will be performed. Radiological signs should reveal subpleural interstitial densities consistent with the diagnosis of IPF including small fibrocystic lesions suggestive of pulmonary fibrosis, yet not extensive honey-combing with significant traction bronchiectasis. Histological proof of a UIP pattern consistent with IPF is generally required. Therefore, surgical lung biopsy is recommended in all cases, if possible (see also Figure 2 below). If a patient does not agree to open lung biopsy, and if the Study Board unanimously agrees that radiological appearance, the pa-tient’s history and the lung function tests are consistent with the clinical diagnosis of IPF, surgical bi-opsy may be omitted. If an open lung biopsy is performed, the histology from those lesions must be consistent with the pattern of UIP. In addition, transbronchial biopsies will be taken from affected pulmonary areas for the initial molecu-lar assessment of the transcription of IFN-, TNF-, TGF-1 and CTGF genes by quantitative RT-PCR. Morover, bronchoalveolar lavage samples will be taken in search for infectious agents. Transbronchial biopsy and bronchoalveolar lavage will be performed at day 0 in every patient for col-lection of study materials as well as on month 12 for sequential assessment of the specimens. Laboratory testing in serum blood samples must exclude collagen vascular diseases or any other sys-temic immune disorders. In total, 30 patients with this condition will be included into the trial.
The following description of patients applies for the screening process in Group B: Patients with proven diagnosis of IPF should present themselves with repeated episodes of coughing, yet not with an acute infectious exacerbation at the time of investigation. Radiological assessment should reveal honeycombing both in different lobes which may also combine subpleural and central regions of each lobe, and may also demonstrate the combination of a reticular pattern with some ground-glass opacities with unequal distribution within the lungs. As it is likely that these radiological features will not allow for an unambiguous diagnosis, histological assessment from the lesions by surgical lung biopsy is required in every case. The findings must be consistent with features of UIP or UIP plus NSIP. PFT should demonstrate a restrictive ventilation pattern. Total lung capacity [TLC] should be between 60 and 80 percent of predicted value, IVC should be comparatively reduced; FVC usually lower than IVC. PFT should not demonstrate significant overinflation of the lungs or significant bronchial obstruction. Measurement of blood gas concentration on controlled physical exertion must be consistent with a reduced pulmonary diffusion capacity. Laboratory testing in serum blood samples must exclude collagen vascular diseases or any other sys-temic immune disorders. In total, 30 patients with this condition will be included into the trial. 6. Diagnostic process and clinical staging Clinical history, physical and radiological findings as well as the results of histological assessment will be reviewed by an expert panel (RESOLVE’s Study Review Board) in accordance with the diagnostic pro-cedure depicted in Figure 2.
There is presently no established treatment for IPF. Given the safety of the treatment with interferon gamma 1b and the considerable experience with the drug, interferon gamma 1b will be used in those patients that fulfill the inclusion criteria described in point 4 and 5. 2. Hypothesis (1)
Condition 1 represents almost exclusively the pathology of pure fibroproliferative repair, and condition 2 most likely the combination of fibroproliferative repair and chronically activate bron-chial inflammation. As a result of the currently late diagnosis of IPF, condition 2 accounts for the majority of patients.
Sequential analysis of whole genome transcriptome and proteome in these two clinical condi-tions over a period of at least 12 months will allow for the identification of relevant mediators of fibroproliferative repair as well as for an assessment of the impact of mediators of chronic bron-chial inflammation on the process of fibroproliferative repair.
By employing an experimental treatment with interferon gamma 1b to 12 patients with condition 1, additional characterization of functionally relevant mediators of fibroproliferative repair as targets for further pharmaceutical development will be possible.
3. Methods Predefined diagnostic criteria used as clinical classifiers for Group A Patients with ‘lone’ IPF (i.e. UIP/IPF without signs of bronchitis) are usually 60-80 years of age and demonstrate a comparatively ‘limited’ form of the disease characterized by the following features:
histological proof of UIP lesions in surgical lung biopsies
radiological signs of subpleural fibrosis
usually no extensive honeycombing in CT scans
usually no significant traction bronchiectasis in CT scans
no signs of widespread ground-glass changes in CT scans
inspiratory vital capacity (IVC) between 70 and 80 percent and higher, sometimes even within nor-mal range
reduction of pulmonary gas exchange, particularly on physical exertion
no symptoms compatible with chronic or acute bronchitis, such as coughing and signs of limitation of expiratory air flow including the ratio of IVC/forced expiratory vital capacity (FVC).
Predefined diagnostic criteria used as clinical classifiers for Group B Patients with IPF complicated by chronic bronchitis, either acquired during the last three years or pre-existing according to the patient’s history: These patients are usually 55-70 years of age and present themselves frequently with a more advanced form of the disease characterized by the following fea-tures:
histological proof of UIP lesions in surgical lung biopsies
signs of traction bronchiectasis in CT scans
clear radiological signs of subpleural fibrosis in at least two lobes of the same lung
inspiratory vital capacity (IVC) between 60 and 80 percent
reduction of pulmonary gas exchange, particularly on physical exertion
symptoms compatible with chronic bronchitis, such as coughing and signs of limitation of expira-tory air flow including the ratio of IVC/forced expiratory vital capacity (FVC).
Patients with IPF/UIP without signs of bronchial disease
histological proof of UIP in surgical lung biopsies or unanimous vote of the
radiological signs suggestive of UIP/IPF
minor radiological signs of honeycombing,
no response to glucocorticoid treatment
no signs of bronchial or systemic inflammation
no transcription of IFN- and TNF- genes
Patients with IPF/UIP with signs of bronchial disease
histological proof of UIP in surgical lung biopsies or unanimous vote for IPF by the study board
radiological signs suggestive of UIP (and possibly also I-NSIP)
clinical symptoms consistent with active bronchitis, or history of chronic bronchitis,
5. Recruitment and characterization of patients It is expected that patients assigned to Group B can be found in specialized pulmonary centers. But, as in Group A, it will be the specific aim of RESOLVE’s study policy to enforce the search for these patients by implementation of local specialists. Patients for Group A may be also found in specialized centers, but will probably be less frequently found in hospitals, as patients at this stage of their disease usually do not have experienced a signifi-cant reduction of their ventilation capacity causing the feeling of intensified dyspnea on exertion. These patients present themselves mostly with symptoms which are not immediately suggestive of pulmonary fibrosis, such as weakness and fatigue with an increase of symptoms over a period of sev-eral months. Notably, this feeling of weakness and/or fatigue is usually attributed by both the patients and also many physicians to increased age or cardiocirculatory comorbidities, respectively. As a result, the recruitment of patients for Group A may pose a problem. In our experience, these pa-tients are mostly seen in outpatient clinics or in ambulatory units, and sometimes by general practitio-ners, as well as by residential specialists in internal medicine, cardiology, and respiratory medicine. The recruitment procedure should thus involve those practitioners and specialists who have repeatedly
send patients to the specialized centers participating in the study. It will be a specific aim of RESOLVE’s study policy to enforce the search for these patients by implementation of local specialists. Screening process in Group A (see also fig. 2): Inclusion into Group A is possible for elderly people (55-80 years of age) who feel physically less capa-ble than they used to do 1 or 2 years ago, but lack any evidence of consuming, cardiac or psychic dis-order (particularly significant depression) at first sight. Patients presenting themselves with these clini-cal signs will then undergo physical exercise testing in the participating centers. The exercise test will be combined with a measurement of arterial blood gas concentration at rest and during physical exer-tion, as reduced pulmonary gas exchange is the first functional sign of UIP/IPF in this group of pa-tients. Measurement of blood gases on controlled physical exertion must be consistent with reduced pulmonary gas diffusion. Pulmonary function test (PFT) may demonstrate a predominantly restrictive ventilatory pattern (usually TLC and IVC > 70%), but can still be normal in group A. However, signs of significant pulmonary em-physema or significant bronchial obstruction should not be present. Once findings of reduced pulmonary gas exchange are confirmed, complete pulmonary function test-ing and CT scan of the lung including high resolution mode will be performed. Radiological signs should reveal subpleural interstitial densities consistent with the diagnosis of IPF including small fibrocystic lesions suggestive of pulmonary fibrosis, yet not extensive honey-combing with significant traction bronchiectasis. Histological proof of a UIP pattern consistent with IPF is generally required. Therefore, surgical lung biopsy is recommended in all cases, if possible (see also Figure 2 below). If a patient does not agree to open lung biopsy, and if the Study Board unanimously agrees that radiological appearance, the pa-tient’s history and the lung function tests are consistent with the clinical diagnosis of IPF, surgical bi-opsy may be omitted. If an open lung biopsy is performed, the histology from those lesions must be consistent with the pattern of UIP. In addition, transbronchial biopsies will be taken from affected pulmonary areas for the initial molecu-lar assessment of the transcription of IFN-, TNF-, TGF-1 and CTGF genes by quantitative RT-PCR. Morover, bronchoalveolar lavage samples will be taken in search for infectious agents. Transbronchial biopsy and bronchoalveolar lavage will be performed at day 0 in every patient for col-lection of study materials as well as on month 12 for sequential assessment of the specimens. Laboratory testing in serum blood samples must exclude collagen vascular diseases or any other sys-temic immune disorders. In total, 30 patients with this condition will be included into the trial.
The following description of patients applies for the screening process in Group B: Patients with proven diagnosis of IPF should present themselves with repeated episodes of coughing, yet not with an acute infectious exacerbation at the time of investigation. Radiological assessment should reveal honeycombing both in different lobes which may also combine subpleural and central regions of each lobe, and may also demonstrate the combination of a reticular pattern with some ground-glass opacities with unequal distribution within the lungs. As it is likely that these radiological features will not allow for an unambiguous diagnosis, histological assessment from the lesions by surgical lung biopsy is required in every case. The findings must be consistent with features of UIP or UIP plus NSIP. PFT should demonstrate a restrictive ventilation pattern. Total lung capacity [TLC] should be between 60 and 80 percent of predicted value, IVC should be comparatively reduced; FVC usually lower than IVC. PFT should not demonstrate significant overinflation of the lungs or significant bronchial obstruction. Measurement of blood gas concentration on controlled physical exertion must be consistent with a reduced pulmonary diffusion capacity. Laboratory testing in serum blood samples must exclude collagen vascular diseases or any other sys-temic immune disorders. In total, 30 patients with this condition will be included into the trial. 6. Diagnostic process and clinical staging Clinical history, physical and radiological findings as well as the results of histological assessment will be reviewed by an expert panel (RESOLVE’s Study Review Board) in accordance with the diagnostic pro-cedure depicted in Figure 2.
 Figure 2. Transcription analysis has been implemented into the screening process due to the improvement of functional diagnostics, particularly with regard the exclusion of pro-inflammatory conditions in Group A. Patients, who agree to surgical lung biopsy, and who demonstrate histological features of either UIP or UIP/ NSIP will be included into the study following informed consent. In contrast to all previous phase III trials, we will, in addition to patient’s history, clinical findings, and histology take advantage of the assessment of gene transcription within the diagnostic process. Patients will only be eligible for inclusion into group A, and particularly for the treatment with inter-feron gamma 1b, if the transcription of the IFN- and TNF- genes is either minimal or undetectable by quantitative PCR technique, and if increased gene transcription for TGF-1 and/or CTGF is observed either from material from the open lung biopsy or from transbronchial biopsies obtained from the le-sions identified by CT scan. The results of every patient wil be made anonymous and reported to the central diagnostic panel for final assessment via RESOLVE’s data exchange system. This decision must be given in written form prior to the inclusion of the patient. 7. Number of Patients and Study Sites Significant decrease of lung function over a period of 12 months has been observed in a group of 49 patients with IIP 12. This observation serves as the basis for the overall estimate of the number of pa-tients in this study. Given the currently low prevalence of IPF, it is thus planned to include a total of 60 patients with IPF into the study. In order to ensure a rapid screening process, the participation of 8 specialized centers in Europe is intended (Berne, CH; Dublin, IRL; Edinburgh, UK; Lublin, PL; Parma, I; Vienna, A; Zabrze, PL; Zurich, CH). If necessary, additional study sites will be included.
8. Description of run-in phase and study-related investigations All patients are assigned to a 2-month run-in period during with an individual oral glucocorticoid treatment (prednisolone 0.5 mg/kg). The effect of this treatment will be tested by complete pulmonary function testing prior to the start of the study. In case of lack of improvement, the patients will be as-signed to the study. The patients will be seen at 4 time points: at a pre-assessment visit, and at visits 1-3 within the obser-vational period. The total observational period will be 12 months. Table 1: Investigations related to the study
Those patients presenting themselves with the clinical condition 1 (IPF without bronchitis) will be as-signed to group A. The total number of patients in group A will be 30. 12 patients of Group A will receive interferon gamma 1b three-times per week subcutaneously at a dose escalating from 20 µg to a maximum of 80 µg over a period of 4 weeks (week 1: 3-times 20 µg; week 2: 3-times 40 µg; week 3: 3-times 60 µg; week 4: 3-times 80 µg). In case of stability or im-provement over a period of three months (Safety Phase), the 12 patients wil be treated for the whole observational period of 12 months. This decision will be made for every patient by the independent Study Review Board. In case of an appearance of symptoms of bronchitis (sputum production, in-creased breathlessness, fatigue and muscle or joint pain, obstructive ventilation pattern in PFT) the treatment will be stopped, and the patients will be assigned to group B. 48 Patients presenting with clinical condition 2 (Group B) will be observed over the study period of 12 months. All patients wil receive the established treatment options for acute infectious exacerbations. Considering the possible functional impact of bronchitis on the overall disease, all patients will daily measure twice the peak expiratory flow via a Peak-Flow protocol that is provided electronically by the participating centers. The patients will return this protocol on a monthly base throughout the study period. Complete assessment by lung function will be done on three visits during the study period.
Histological, molecular, and biochemical assessment for target identification Study materials Assessment of whole genome transcriptomics and full scale proteomics will be performed at visit 1 and 3 of the trial (month 0 and month 12). The measurements will be based on a) open lung biopsy (if suit-able), b) on transbronchial biopsies (TbX) taken during broncoscopy, c) cells recovered from bron-choalveolar lavage (BAL). In addition, BAL fluid will be stored and used for detection of proteins and lipids. Materials for DNA methylation analysis will be obtained from whole blood samples. These as-sessments will be performed independently by Partner 10 of the RESOLVE project (Austrian Research Centers, Seibersdorf, Austria; ARC). Immunohistochemistry will be performed at the Department of Pathology of the University of Verona, Italy (Partner 4 of the RESOLVE project), both in surgical lung biopsies as well as in transbronchial lung biopsies. Additionally, ARC shall establish primary cell cultures (epithelial cells and fibroblasts) either from the open lung biopsies or from the TbX in order to provide suitable in vitro test systems for the validation of targets. Blinding and Dealing with personalized information All information, i.e. clinical data and study materials obtained at visit 1-3, will be made anonymous be allocating them to the study numbers given consecutively to each patient included in the study. These study numbers will serve as a unique identifier for each patient. All personal information (particularly the clinical classifiers used for stratification of the patients) is kept at the clinical centers conducting the study and will not be made available to the units that per-form the scientific assessments. Clinical classifiers and results of the assessment of study materials identified by the study number will be made accessible to the independent mathematical evaluation unit (Partners 2, Integromics S.A., Granada, Spain, and Partner 13, Centro Nacional de Biotechnologica, Madrid, Spain) and to the partici-pants conducting the mathematical modeling calculations (Partner 3, Politecnico de Torino). A data exchange system (LIMS) has been set up providing the platform for complte and rapid exchange of the digitalized data. Data entry and correctness of data are controlled by an independent monitor (Raba & Morgenstern Pharma Consulting, Oberhaching, Germany). All data are stored on predefined CMEs on a secure server system provided by Partner 2. Each individual accession to the database is continuously monitored by individual accession number, date and time.
9. Safety measures Safety will be assessed by RESOLVE’s Study Review Board. With respect to the present study, the follow-ing clinical events will be closely monitored: 1. No improvement after immunosuppressive treatment with glucocorticosteroids. 2. No signs of bronchitis as well as no transcription of IFN- and TNF- genes in the subgroup of pa-
tients who will receive treatment with interferon gamma 1b.
3. Side-effects indicative of bronchitis after treatment with interferon gamma 1b within the first three
months of the trial (Safety Phase) in the subgroup receiving interferon gamma 1b.
4. Signs indicative of bronchial disease in the Peak Flow protocol during the Safety Phase. 10. References 1. American Thoracic Society/European Respiratory Society. International Multidisciplinary Consen-
sus Classification of the Idiopathic Interstitial Pneumonias. Am J Respir Crit Care Med 2002; 165: 277–304.
2. Antoniou KM, Alexandrakis MG, Siafakas NM, Bouros D: Cytokine network in the pathogenesis of
idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 2005; 22(2): 91-104.
3. Swigris JJ, Kuschner WG, Kelsey JL, Gould MK. Idiopathic pulmonary fibrosis: challenges and op-
portunities for the clinician and investigator. Chest 2005; 127: 275–283.
4. Travis WD, King TE, Bateman ED, Lynch DA, Capron F, Center D, Colby TW, Cordier J-F, Du Bois
RM, Grenier GJ, et al. American Thoracic Society/European Respiratory Society International mul-tidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2002; 165: 277–304.
5. Nicholson AG, Colby TV, du Bois RM, Hansell DM, Wells AU. The prognostic significance of the
histologic pattern of interstitial pneumonia in patients presenting with the clinical entity of cryp-togenic fibrosing alveolitis. Am J Respir Crit Care Med 2000; 162: 2213–2217.
6. Flaherty KR, Travis WD, Colby TV, Toews GB, Kazerooni EA, Gross BH, Jain A, Strawderman RL,
Flint A, Lynch JP, et al. Histopathologic variability in usual and nonspecific interstitial pneumonias. Am J Respir Crit Care Med 2001; 164: 1722–1727.
7. Steele MP, Speer MC, Loyd JE, Brown KK, Herron A, Slifer SH, Burch LH, Wahidi MM, Phillips JA III,
Sporn TA, et al. The clinical and pathologic features of familial interstitial pneumonia (FIP). Am J Respir Crit Care Med 2005; 172: 1146–1152.
8. Katzenstein AL, Zisman DA, Litzky LA, Nguyen BT, Kotloff RM. Usual interstitial pneumonia: his-
tologic study of biopsy and explant specimens. Am J Surg Pathol 2002; 26: 1567–1577.
9. Wang XM, Zhang Y, Kim HP, Zhou Z, Feghali-Bostwick CA et al. Caveolin-1: a critical regulator of
lung fibrosis in idiopathic pulmonary fibrosis. J Exp Med 203: 2895–2906 (2006)
10. Strieter RM: Pathogenesis and Natural History of Usual Interstitial Pneumonia. Chest 2005; 128:
11. Selman M, Pardo A, Barrera L, Estrada A, Watson SR, Wilson K, Aziz N, Kaminski N, Zlotnik A: Gene
expression profiles distinguish idiopathic pulmonary fibrosis from hypersensitivity pneumonitis. Am J Respir Crit Care Med 2006; 173(2): 188-198.
12. Kaminski, N, Rosas IO: Gene Expression Profiling as a Window into Idiopathic Pulmonary Fibrosis
Pathogenesis. Proc Am Thorac Soc 2006; 3: 339–344.
13. Golec M, Lambers C, Hofbauer E, Geleff S, et al. Assessment of gene transcription demonstrates
connection with the clinical course of idiopathic interstitial pneumonia. Respiration. Online ver-sion: DOI: 10.1159/000137511 (2008).
14. Du Bois R, King TE. Challenges in pulmonary fibrosis. The NSIP/UIP debate. Thorax 62, 1008-
15. American Thoracic Society/European Respiratory Society. International Multidisciplinary Consen-
sus Classification of the Idiopathic Interstitial Pneumonias. Am J Respir Crit Care Med 165: 277–304 (2002).
Figure 2. Transcription analysis has been implemented into the screening process due to the improvement of functional diagnostics, particularly with regard the exclusion of pro-inflammatory conditions in Group A. Patients, who agree to surgical lung biopsy, and who demonstrate histological features of either UIP or UIP/ NSIP will be included into the study following informed consent. In contrast to all previous phase III trials, we will, in addition to patient’s history, clinical findings, and histology take advantage of the assessment of gene transcription within the diagnostic process. Patients will only be eligible for inclusion into group A, and particularly for the treatment with inter-feron gamma 1b, if the transcription of the IFN- and TNF- genes is either minimal or undetectable by quantitative PCR technique, and if increased gene transcription for TGF-1 and/or CTGF is observed either from material from the open lung biopsy or from transbronchial biopsies obtained from the le-sions identified by CT scan. The results of every patient wil be made anonymous and reported to the central diagnostic panel for final assessment via RESOLVE’s data exchange system. This decision must be given in written form prior to the inclusion of the patient. 7. Number of Patients and Study Sites Significant decrease of lung function over a period of 12 months has been observed in a group of 49 patients with IIP 12. This observation serves as the basis for the overall estimate of the number of pa-tients in this study. Given the currently low prevalence of IPF, it is thus planned to include a total of 60 patients with IPF into the study. In order to ensure a rapid screening process, the participation of 8 specialized centers in Europe is intended (Berne, CH; Dublin, IRL; Edinburgh, UK; Lublin, PL; Parma, I; Vienna, A; Zabrze, PL; Zurich, CH). If necessary, additional study sites will be included.
8. Description of run-in phase and study-related investigations All patients are assigned to a 2-month run-in period during with an individual oral glucocorticoid treatment (prednisolone 0.5 mg/kg). The effect of this treatment will be tested by complete pulmonary function testing prior to the start of the study. In case of lack of improvement, the patients will be as-signed to the study. The patients will be seen at 4 time points: at a pre-assessment visit, and at visits 1-3 within the obser-vational period. The total observational period will be 12 months. Table 1: Investigations related to the study
Those patients presenting themselves with the clinical condition 1 (IPF without bronchitis) will be as-signed to group A. The total number of patients in group A will be 30. 12 patients of Group A will receive interferon gamma 1b three-times per week subcutaneously at a dose escalating from 20 µg to a maximum of 80 µg over a period of 4 weeks (week 1: 3-times 20 µg; week 2: 3-times 40 µg; week 3: 3-times 60 µg; week 4: 3-times 80 µg). In case of stability or im-provement over a period of three months (Safety Phase), the 12 patients wil be treated for the whole observational period of 12 months. This decision will be made for every patient by the independent Study Review Board. In case of an appearance of symptoms of bronchitis (sputum production, in-creased breathlessness, fatigue and muscle or joint pain, obstructive ventilation pattern in PFT) the treatment will be stopped, and the patients will be assigned to group B. 48 Patients presenting with clinical condition 2 (Group B) will be observed over the study period of 12 months. All patients wil receive the established treatment options for acute infectious exacerbations. Considering the possible functional impact of bronchitis on the overall disease, all patients will daily measure twice the peak expiratory flow via a Peak-Flow protocol that is provided electronically by the participating centers. The patients will return this protocol on a monthly base throughout the study period. Complete assessment by lung function will be done on three visits during the study period.
Histological, molecular, and biochemical assessment for target identification Study materials Assessment of whole genome transcriptomics and full scale proteomics will be performed at visit 1 and 3 of the trial (month 0 and month 12). The measurements will be based on a) open lung biopsy (if suit-able), b) on transbronchial biopsies (TbX) taken during broncoscopy, c) cells recovered from bron-choalveolar lavage (BAL). In addition, BAL fluid will be stored and used for detection of proteins and lipids. Materials for DNA methylation analysis will be obtained from whole blood samples. These as-sessments will be performed independently by Partner 10 of the RESOLVE project (Austrian Research Centers, Seibersdorf, Austria; ARC). Immunohistochemistry will be performed at the Department of Pathology of the University of Verona, Italy (Partner 4 of the RESOLVE project), both in surgical lung biopsies as well as in transbronchial lung biopsies. Additionally, ARC shall establish primary cell cultures (epithelial cells and fibroblasts) either from the open lung biopsies or from the TbX in order to provide suitable in vitro test systems for the validation of targets. Blinding and Dealing with personalized information All information, i.e. clinical data and study materials obtained at visit 1-3, will be made anonymous be allocating them to the study numbers given consecutively to each patient included in the study. These study numbers will serve as a unique identifier for each patient. All personal information (particularly the clinical classifiers used for stratification of the patients) is kept at the clinical centers conducting the study and will not be made available to the units that per-form the scientific assessments. Clinical classifiers and results of the assessment of study materials identified by the study number will be made accessible to the independent mathematical evaluation unit (Partners 2, Integromics S.A., Granada, Spain, and Partner 13, Centro Nacional de Biotechnologica, Madrid, Spain) and to the partici-pants conducting the mathematical modeling calculations (Partner 3, Politecnico de Torino). A data exchange system (LIMS) has been set up providing the platform for complte and rapid exchange of the digitalized data. Data entry and correctness of data are controlled by an independent monitor (Raba & Morgenstern Pharma Consulting, Oberhaching, Germany). All data are stored on predefined CMEs on a secure server system provided by Partner 2. Each individual accession to the database is continuously monitored by individual accession number, date and time.
9. Safety measures Safety will be assessed by RESOLVE’s Study Review Board. With respect to the present study, the follow-ing clinical events will be closely monitored: 1. No improvement after immunosuppressive treatment with glucocorticosteroids. 2. No signs of bronchitis as well as no transcription of IFN- and TNF- genes in the subgroup of pa-
tients who will receive treatment with interferon gamma 1b.
3. Side-effects indicative of bronchitis after treatment with interferon gamma 1b within the first three
months of the trial (Safety Phase) in the subgroup receiving interferon gamma 1b.
4. Signs indicative of bronchial disease in the Peak Flow protocol during the Safety Phase. 10. References 1. American Thoracic Society/European Respiratory Society. International Multidisciplinary Consen-
sus Classification of the Idiopathic Interstitial Pneumonias. Am J Respir Crit Care Med 2002; 165: 277–304.
2. Antoniou KM, Alexandrakis MG, Siafakas NM, Bouros D: Cytokine network in the pathogenesis of
idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 2005; 22(2): 91-104.
3. Swigris JJ, Kuschner WG, Kelsey JL, Gould MK. Idiopathic pulmonary fibrosis: challenges and op-
portunities for the clinician and investigator. Chest 2005; 127: 275–283.
4. Travis WD, King TE, Bateman ED, Lynch DA, Capron F, Center D, Colby TW, Cordier J-F, Du Bois
RM, Grenier GJ, et al. American Thoracic Society/European Respiratory Society International mul-tidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2002; 165: 277–304.
5. Nicholson AG, Colby TV, du Bois RM, Hansell DM, Wells AU. The prognostic significance of the
histologic pattern of interstitial pneumonia in patients presenting with the clinical entity of cryp-togenic fibrosing alveolitis. Am J Respir Crit Care Med 2000; 162: 2213–2217.
6. Flaherty KR, Travis WD, Colby TV, Toews GB, Kazerooni EA, Gross BH, Jain A, Strawderman RL,
Flint A, Lynch JP, et al. Histopathologic variability in usual and nonspecific interstitial pneumonias. Am J Respir Crit Care Med 2001; 164: 1722–1727.
7. Steele MP, Speer MC, Loyd JE, Brown KK, Herron A, Slifer SH, Burch LH, Wahidi MM, Phillips JA III,
Sporn TA, et al. The clinical and pathologic features of familial interstitial pneumonia (FIP). Am J Respir Crit Care Med 2005; 172: 1146–1152.
8. Katzenstein AL, Zisman DA, Litzky LA, Nguyen BT, Kotloff RM. Usual interstitial pneumonia: his-
tologic study of biopsy and explant specimens. Am J Surg Pathol 2002; 26: 1567–1577.
9. Wang XM, Zhang Y, Kim HP, Zhou Z, Feghali-Bostwick CA et al. Caveolin-1: a critical regulator of
lung fibrosis in idiopathic pulmonary fibrosis. J Exp Med 203: 2895–2906 (2006)
10. Strieter RM: Pathogenesis and Natural History of Usual Interstitial Pneumonia. Chest 2005; 128:
11. Selman M, Pardo A, Barrera L, Estrada A, Watson SR, Wilson K, Aziz N, Kaminski N, Zlotnik A: Gene
expression profiles distinguish idiopathic pulmonary fibrosis from hypersensitivity pneumonitis. Am J Respir Crit Care Med 2006; 173(2): 188-198.
12. Kaminski, N, Rosas IO: Gene Expression Profiling as a Window into Idiopathic Pulmonary Fibrosis
Pathogenesis. Proc Am Thorac Soc 2006; 3: 339–344.
13. Golec M, Lambers C, Hofbauer E, Geleff S, et al. Assessment of gene transcription demonstrates
connection with the clinical course of idiopathic interstitial pneumonia. Respiration. Online ver-sion: DOI: 10.1159/000137511 (2008).
14. Du Bois R, King TE. Challenges in pulmonary fibrosis. The NSIP/UIP debate. Thorax 62, 1008-
15. American Thoracic Society/European Respiratory Society. International Multidisciplinary Consen-
sus Classification of the Idiopathic Interstitial Pneumonias. Am J Respir Crit Care Med 165: 277–304 (2002).